
A kaleidoscopic view of extracellular vesicles in lysosomal storage disorders
 0
0
Abstract
Extracellular vesicles (EVs) are a heterogeneous population of stable lipid membrane particles that play a critical role in the regulation of numerous physiological and pathological processes. EV cargo, which includes lipids, proteins, and RNAs including miRNAs, is affected by the metabolic status of the parental cell. Concordantly, abnormalities in the autophagic-endolysosomal pathway, as seen in lysosomal storage disorders (LSDs), can affect EV release as well as EV cargo. LSDs are a group of over 70 inheritable diseases, characterized by lysosomal dysfunction and gradual accumulation of undigested molecules. LSDs are caused by single gene mutations that lead to a deficiency of a lysosomal protein or lipid. Lysosomal dysfunction sets off a cascade of alterations in the endolysosomal pathway that can affect autophagy and alter calcium homeostasis, leading to energy imbalance, oxidative stress, and apoptosis. The pathophysiology of these diseases is very heterogenous, complex, and currently incompletely understood. LSDs lead to progressive multisystemic symptoms that often include neurological deficits. In this review, a kaleidoscopic overview will be given on the roles of EVs in LSDs, from their contribution to pathology and diagnostics to their role as drug delivery vehicles. Furthermore, EV cargo and surface engineering strategies will be discussed to show the potential of EVs in future LSD treatment, both in the context of enzyme replacement therapy, as well as future gene editing strategies like CRISPR/Cas. The use of engineered EVs as drug delivery vehicles may mask therapeutic cargo from the immune system and protect it from degradation, improving circulation time and targeted delivery.
Keywords
INTRODUCTION
Lysosomal storage disorders (LSDs) include over 70 rare heritable (inborn) errors of metabolism. In LSDs, a single gene mutation causes deficiency of a lysosomal protein or lipid, which leads to gradual accumulation of undigested macromolecules and negatively affects lysosomal function. Because lysosomal hydrolases are involved in the stepwise degradation, primary substrate accumulation in lysosomes gradually leads to secondary accumulation of upstream substrates within the same catabolic pathway. The accumulation leads to enlargement of the organelle and functional inhibition of genetically correct enzymes and proteases which further limits their processing abilities[1-3]. This sets off a cascade of alterations in the endolysosomal pathway due to impaired fusion that can affect autophagy, but also in other organelles including the endoplasmic reticulum and mitochondria[4]. This may further lead to altered calcium homeostasis, oxidative stress, energy imbalance, and apoptosis[5,6]. The degree of the effect on these cellular processes highly depends on the type of LSD and its specific genetic mutation[7]. The complex interplay of the cellular pathways is shown in Figure 1. The cellular impairments seen in LSDs can lead to cell death and cause progressive multisystemic symptoms that often include visceromegaly, bone abnormalities, cardio-respiratory problems, and neurodegeneration[8,9].
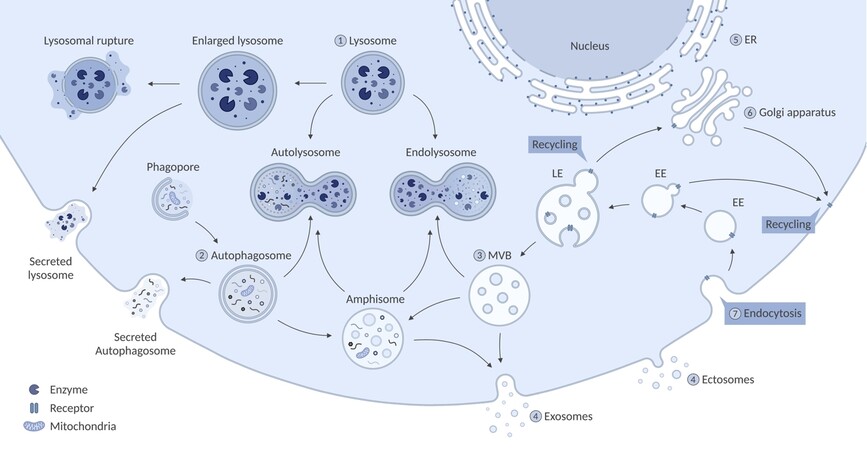
Figure 1. A schematic illustration of different pathways affected in LSDs. (1) In LSDs, a single gene mutation causes the deficiency of a lysosomal protein or lipid. The defect leads to gradual accumulation of macromolecules, inhibition of other lysosomal components, and enlargement of the lysosome which can be followed by lysosomal rupture and secretion. Furthermore, dysfunction of the lysosome and its altered calcium (Ca2+) homeostasis leads to impaired fusion with cellular structures including autophagosomes and endosomes/multivesicular bodies (MVBs). (2) The accumulation of autophagosomes can potentially lead to secretion of the organelle to clear its obstruction. Additionally, autophagy is downregulated by hyperactivation of mTOR in some LSDs (MPS-I, MPS-VI, MPS-VII, and Gaucher Disease). Subsequently, impaired or inhibited autophagy results in mitochondrial dysfunction that can lead to energy imbalance, apoptosis, and inflammation. (3) There is a balance between degradation and EV release, which is highly dependent on the faith of MVBs. (4) Impaired fusion of MVBs with the lysosome can lead to fusion of MVBs with the plasma membrane, which causes increased secretion of EVs (Niemann-Pick type C, Sialidosis, Krabbe and Fabry Disease). Hence, inhibition of autophagy and subsequential increase of EV secretion might be an alternative route for LSD cells to clear obstruction of the accumulating molecules to naturally improve the cellular phenotype. (5) In multiple sphingolipid storage disorders (Gaucher Disease, GM1 and GM2 Gangliosidoses, and Niemann-Pick type A) Endoplasmic reticulum (ER) calcium homeostasis is perturbed, which leads to elevated Ca2+ in the cytosol. (6) Dysfunction of the Golgi apparatus has been reported for most lipid storage disorders and MPS IIIB. This includes altered morphology and accumulation of large storage bodies.
Around 70% of the LSDs are enzyme deficiencies and can be further subclassified based on their storage material, such as Glycoproteinoses, Mucopolysaccharidoses, and Lipidoses. Other subgroups of LSDs include deficiencies in integral membrane proteins, transporters and proteins involved in trafficking, regulation, or post-translational modification. Despite the classification, LSDs are clinically very heterogeneous, even within the same subtype. The symptoms depend on the specific genetic mutation and to which degree this affects the lysosomal protein or lipid, as well as the spatial and temporal accumulation of macromolecules[7]. While individual LSDs are rare, collectively, they have an estimated incidence of 1 in 5000-8000[10-12]. All LSDs that will be described in this review are listed in Table 1.
Described Lysosomal Storage Disorders
| Disease (gene) Eponyms and/or types | Primary deficiency | Accumulated molecules | Approved therapies |
Sphingolipidoses | |||
| Fabry Diseasea (GLA) | α- Galactosidase A | Globotriaosylceramide | ERT and CT |
| Farber Lipogranulomatosisa (ASAH1) | Acid ceramidase | Ceramide | S/S |
| Gaucher Diseasea (GBA) Type I, IIb, IIIb, perinatal lethal formb and cardiovascular[13]c | β Glucocerebrosidase/β- glucosidase | Glucocerebroside/ glucosylsphingosine | ERT and SRT (types I, II and III), none (perinatal lethal form) |
| GM2 Gangliosidosis Tay-Sachs Diseasec (HEXA) | β- Hexosaminidase | GM2 ganglioside, glycosphingolipids and oligosaccharides | S/S |
| Globoid Cell Leukodystrophya,b (GALC) Krabbe Disease | Galactosylceramidase | Galactocerebroside and psychosine | HSCT (infantile onset) and S/S |
| Metachromatic Leukodystrophya,b (ARSA and PSAP) MLD | Arylsulfatase A Sphingolipid activator protein SAP-B | Sulfatides | HSCT |
| Niemann-Pick Diseasea (SMPD1) Type Ab and B | Sphingomyelin Phosphodiesterase | Sphingomyelin | S/S |
Mucopolysaccharidosis (MPS) | |||
| MPS IIIa,b,d III-A Sanfilippo A (SGSH) III-C Sanfilippo C (HGSNAT) | N- Sulfoglucosamine sulfohydrolase Heparan- α-glucosaminide- N- acetyltransferase | HS (KS) | S/S |
| MPS IVa IV-A Morquio syndrome A (GALNS) | N-acetylgalactosamine- 6-sulfatase | KS, C6S | ERT (IV-A) and S/S (IV-B) |
| MPS VIIa,b (GUSB) Sly Disease | β- Glucuronidase | DS, HS, C4S, C6S (KS) | ERT |
Glycogen Storage Disorders (GSD) | |||
| GSD IIa (GAA) Pompe Disease | Lysosomal α- glucosidase/acid maltase | Glycogen | ERT |
Glycoproteinoses | |||
| Sialidosisa (NEU1) Type I, cherry-red spot myoclonus Type IIb, Mucolipidosis I | Neuraminidase-1 | Sialylated oligosaccharides and glycopeptides Sialylated oligosaccharides and glycopeptides, LAMP1 and amyloid precursor protein | S/S |
Integral Membrane Protein Disorders | |||
| Cystinosis (CTNS) | Cystinosin | Cystine | SMT |
| Niemann-Pick Disease C1 (NPC1) C2b (NPC2) | NPC intracellular cholesterol transporter 1 NPC intracellular cholesterol transporter 2 | Cholesterol and sphingolipids | SRT |
Neuronal Ceroid Lipofuscinoses (NCL or CLN), Batten Disease | |||
| CLN2b (TPP1) Jansky-Bielschowsky Disease | Tripeptidyl peptidase 1 | Subunit c of mitochondrial ATP synthase | ERT |
To date, the most established treatment option for LSDs is enzyme replacement therapy (ERT)[16]. ERT is available for several enzyme deficiencies, where it can improve clinical symptoms and slow down disease progression. Unfortunately, ERT comes with serious limitations that include the development of neutralizing antibodies against the enzyme, low plasma half-life, inability to cross the blood-brain barrier (BBB), and high costs. Furthermore, patients treated with ERT are left with residual disease due to its poor biodistribution, which fails to effectively target certain organs[17-19]. Besides ERT, hematopoietic stem cell transplantation (HSCT) is available for a very limited group of LSDs, including MPS-IH, MPS-II, α-Mannosidosis, MLD, and Krabbe Disease. To improve its effects and survival of patients, early diagnosis and early treatment with HSCT are critical. The effect of stem cell transplantation is based on the production of the missing enzyme by the healthy donor cells that will allow cross-correction[20-25]. However, HSCT is not fully curative and unable to completely halt disease in hard-to-reach tissues. This is often the case for connective tissue, resulting in progression of skeletal malformations, corneal clouding, valvular dysfunction, as well as inconsistent results for joint, tendon, and ligament stiffness[15]. Several other novel strategies have been developed over the years, including substrate reduction therapy (SRT), pharmacological chaperone therapy (PCT), and gene therapy[26-28]. For most LSDs, the only solution remains symptomatic treatment and supportive care[7]. A cure that will be more broadly applicable to treat more LSDs, including those with severely affected neurological manifestations, has yet to be discovered.
Post-treatment residual disease in connective tissues has drawn attention towards multipotent mesenchymal stem cells (MSCs), which have the ability to differentiate into multiple lineages of bone, cartilage, and fat. Moreover, MSCs are characterized by their intrinsic self-renewal capacity and their capability of immunomodulation. Their transplantation is widely tested in clinical trials and considered safe[29,30]. In comparison to hematopoietic stem cells (HSC), MSCs secrete significantly higher levels of mucopolysaccharidosis (MPS)-related enzymes[31]. Thus, MSCs might be able to improve skeletal malformations and neurological deficits in LSD patients at an early stage of disease development[29,32]. Several studies with MSCs derived from various tissue sources have been performed in mice models for different LSDs. In these studies, improvement in motor function, decreased inflammation and apoptosis were reported post-injection of the cells into different parts of the brain[5]. To date, intravenous injection of MSCs has only been tested on patients with Metachromatic Leukodystrophy (MLD); this showed promising results regarding its safety and improvement of stabilization on neurological manifestations[33,34]. Currently, a clinical trial is assessing the safety of human placental-derived MSCs (phase I) in multiple other LSDs (ID: NCT01586455). However, increasing evidence suggests that the therapeutic efficiency of MSC therapy is not dependent on the engraftment of MSCs or their capability to differentiate after transplantation. Most likely, the mode of action of MSCs relies on their paracrine signaling including extracellular vesicles (EVs) to support surrounding tissues[35].
Over the last two decades, attention has been drawn towards EVs as important mediators of intercellular communication. EVs are a heterogeneous population of stable lipid membrane particles and play a critical role in the regulation of numerous physiological and pathological processes[36]. EVs capture the metabolic status of the cell[37], which qualifies them as potential sources of biomarkers for LSDs. Furthermore, EVs are biocompatible, low immunogenic and can traverse multiple biological barriers. For instance, several studies have indicated that EVs are able to cross the BBB[38,39]. Additionally, bioengineering approaches can modify EVs to load specific cargo or target specific sites[40]. This makes EVs of high interest as both therapeutic agents and drug delivery vehicles, which could open doors to the development of novel treatments for LSDs including those with neurological deficits. In this review, we will provide an overview of the current knowledge and potential of EVs in unraveling LSDs, their diagnostics, treatment, and potential future drug delivery for gene correction strategies. For an overview of the existing synthetic nanotechnology-based approaches that have been explored as delivery vehicles in the treatment of LSDs, we refer the reader to the reviews of Abasolo et al., Schuh et al., and Del Grosso et al.[41,42,43].
THE BIOGENESIS OF EXTRACELLULAR VESICLES
EVs are a heterogeneous population of small lipid membrane vesicles that are secreted by all cells, and have been observed in all body fluids[36]. EVs are conventionally classified into subtypes based on their biogenesis: exosomes and ectosomes. Exosomes (40-140 nm) are a part of the endo-lysosomal system and form through intraluminal vesicle (ILV) formation by inward membrane budding in the late endosome. This process involves the endosomal sorting complexes required for transport (ESCRT) machinery, tetraspanins and lipid-dependent interactions, which ultimately leads to the formation of multivesicular bodies (MVBs)[44]. MVBs can release their intraluminal vesicles upon fusion with the plasma membrane into the extracellular space, after which they are referred to as exosomes. Ectosomes, also referred to as microvesicles
EVs are known for their contribution to cell-to-cell communication by transferring cargo such as RNA, including mRNA and long noncoding RNAs, lipids, and proteins including lysosomal enzymes[48-50]. Over the last decade, it has become clear that EV cargo reflects the status of the secreting cell and its tissue origin[37,51]. Cargo transportation through EVs plays a critical role in the regulation and progression of numerous physiological and pathological processes[36]. The contribution of EVs to pathology has been shown for several neurological disorders that have been associated with LSDs or other pathologies related to endolysosomal dysfunction, such as Parkinson’s Disease (PD)[52], Alzheimer’s Disease[53], and Huntington’s Disease[54,55]. Since these neurological disorders are currently not classified as classical LSDs, describing the endolysosomal involvement in the pathology of these disorders is beyond the scope of this review. For more in-depth information on these diseases, we refer the readers to the reviews of Navarro-Romero et al. and Almeida et al.[56,57]. In these neurological disorders, EVs are thought to contribute to inflammation and seem to sequester and spread the accumulating pathogenic proteins[53,58]. This suggests that EVs may contribute to the altered physiology in LSDs as well[59]. This makes EVs a potentially useful source of information to further determine the altered processes in LSDs as biomarkers for diagnostics and disease progression, including their location[51,60].
As EVs hold the ability to traverse multiple biological barriers and can be modified to avoid uptake by the mononuclear phagocytic system, EVs are potential therapeutic agents and drug delivery vehicles for LSDs, due to their biocompatibility and low immunogenicity[61-63]. Adaptation of EVs through bioengineering approaches could further optimize their delivery potential and therapeutic value by targeted loading of therapeutic components through fusion proteins, or to target EVs to specific tissues[40,64]. This strategy could actively load ERT into EVs, which could reduce neutralizing antibodies against the enzyme, improve its plasma half-life, and facilitate delivery towards the nervous system by crossing the BBB[38]. As such, EVs have the potential to both improve existing treatment and lead to new therapeutic options for LSD patients in the future, including those with severe neurological deficits.
EV RELEASE IN LSDs: A NEW ROUTE IN UNDERSTANDING DISEASE PATHOLOGY
Lysosomes are essential organelles in the degradation of macromolecules delivered by endocytosis or autophagocytosis. The lysosomal characteristic acidic lumen includes over 60 hydrolases that can return molecules to catabolites, thereby allowing recycling of these molecules. These functions allow lysosomes to play a fundamental role in multiple cellular processes including autophagy and its regulation, cellular homeostasis, cell signaling, mitochondrial function and vesicle fusion[65,66]. Several of these processes are also implicated in EV release and uptake, which are both parts of the endolysosomal pathway. As described before, the fusion of MVBs with the plasma membrane leads to EV release. Alternatively, MVBs can fuse with both lysosomes and autophagosomes, leading to degradation of MVBs. Moreover, both studies on ESCRT proteins and autophagy modulators show a tight relationship between autophagy and MVB biogenesis, thereby determining the faith of its ILVs and, subsequently, of EVs[44]. The lysosomal dysfunction in LSDs does not make it difficult to envision its effects on both EV release and uptake, which will be discussed in this section.
Autophagy is a multi-step process where damaged organelles and molecular aggregates are degraded and recycled. The process starts with the formation of autophagosomes that can fuse with early and late endosomal vesicles, such as MVBs, to form amphisomes, to eventually fuse with lysosomes[4,7]. Under homeostatic conditions, cells maintain a constitutive basal level of autophagy; however, the process can be induced in response to cellular and metabolic stress[67]. In several LSDs, it has been shown that lysosomal dysfunction leads to the accumulation of autophagosomes and autophagic substrates, such as damaged mitochondria[4,68,69]. However, accumulation of autophagosomes does not necessarily indicate an increased induction of autophagy as this increase could also be caused by blockage of fusion with the lysosome. Nevertheless, the autophagy pathway in LSDs is disrupted, which could either mean activation or inhibition of autophagy, depending on the LSD type. For instance, in MPS-I, MPS-VI, MPS-VII, and Gaucher Disease, hyperactivation of mTOR, a negative regulator of autophagy, has been shown[69,70]. In this study, mTOR hyperactivation increased phosphorylation of the UVRUG protein, which led to reduced autophagosome maturation and reduced fusion with the lysosome[69]. Moreover, activated mTOR can phosphorylate transcription factor EB (TFEB), which inhibits its translocation to the nucleus and, with that, lysosomal biogenesis and function[8]. Thus, there might be a vicious cycle in play where lysosomal accumulation inhibits the induction of autophagy, perhaps to prevent further accumulation-induced damage. The complex interplay of autophagy and the endolysosomal pathways is shown in Figure 1.
There is a balance between autophagy and EV release, which is highly dependent on the faith of MVBs. It has been shown that in hepatic stellate cells, activation of mTOR signaling induced the release of exosomes by inhibiting autophagy. Furthermore, the activation of mTOR can stimulate release of microvesicles through activation of ROCK1 signaling[71]. The hyperactivation of mTOR in some LSDs could play a vital part in the secretion of EVs by these cells. In a study by Bartolomeo et al., it was shown that mTOR hyperactivity significantly reduced the formation of Phosphatidylinositol-3-phosphate (PI3P)[69]. This may influence EV release considering that PI3P is required in vesicle and (auto)phagosome fusion with lysosomes. As PI3P reduction may limit the fusion between MVBs and autophagosomes, this could potentially shift the faith of MVBs towards fusion with the plasma membrane and secretion of EVs instead, to limit accumulation and maintain homeostasis. Furthermore, PI3P is involved in ESCRT protein recruitment and cargo organization, and serves as a substrate for PI(3,5)P2 formation[72,73]. Therefore, the diminished presence of PI3P through mTOR hyperactivation could also lead to reduced PIP2 formation. The absence of PIP2 has been shown to increase EV secretion[74]. In addition, reduction of PI3P formation promoted secretion of atypical exosome containing undigested lysosomal components in neuronal disorders with lysosomal dysfunction closely related to Niemann-Pick type C[73,75]. A study by Strauss et al. confirmed increased EV release in Niemann-Pick type C cells[76]. Table 2 gives an overview of all studies describing the EV release by LSD cells. In Niemann-Pick type C, loss of function of a late endosomal membrane protein leads to accumulation of free cholesterol and sphingolipids. Strauss et al. showed a flotillin-dependent pathway of EV secretion as an alternative to secrete free cholesterol to ameliorate its storage within cells[76,77]. Moreover, increased EV release has been shown in several enzyme deficiencies, including Sialidosis, Krabbe and Fabry Disease[68,78,79]. Hence, inhibition of autophagy and subsequential increase of EV secretion might be an alternative route for LSD cells to clear obstruction of the accumulating molecules to improve the cellular phenotype.
The secretion of extracellular vesicles in lysosomal storage disorder models
| LSD | Disease model | EV isolation method | Major results | Reference |
| Fabry Disease | In vitro CRISPR/Cas9 altered human embryonic stem cells differentiated into cardiomyocytes | Total Exosome Isolation Reagent (Thermo Fisher Scientific) | > Downregulation of proteins related to EV transportation, secretion, and exocytosis (Rab GTPases: Rab-11, GDIR2, VPS36 and VTI1A) > Impairment of the autophagic flux and protein turnover, which increased reactive oxygen species, apoptosis, and necrosis > Increased production and secretion of exosomes | [68] |
| Farber Lipogranulo-matosis (Partial model) | Asah1fl/fl/Smooth Muscle (SM)cre mice | Centrifugation (4 °C): 300 g, 10 min Filter 0.22 µm filters to 100,000 g, 90 min Washing step 100,000 g, 90 min | > Acid ceramidase (Ac) contributes to the development of arterial medial calcification (AMC) in the aorta and increases osteogenic markers > Decreased colocalization of MVBs with lysosomes and upregulation of sEV markers was seen in coronary arterial smooth muscle cells (CASMCs) of vitamin D treated Asah1fl/fl/SMCre mice; The same effect was observed in phosphate (Pi) treated CASMCs in vitro > Untreated Asah1fl/fl/SMCre CASMCs show increased sEV release, whereas Pi treated > Ac gene deletion remarkably decreased ML-SA1 induced Ca2+ release through TRPML1 channels | [80] |
| Krabbe Disease | In vitro Primary erythrocytes and oligodendrocytes from Twitcher mice-pups | Centrifugation: 100 g, 10 min at RT 21.000 g, 20 min at 4 °C | > Psychosine accumulation introduces focal regions of rigidity in the plasma membrane, which affects curvature and facilitates increased budding and shedding of 0.5-4 μm EVs in erythrocytes and oligodendrocytes | [79] |
| In vivo Twitch mice (GALC-/-) | Centrifugation: 300 g, 10 min (P1) 2.000 g, 10 min (P2) 10,000 g, 30 min (P3) 100,000 g, 90 min (P4) Sucrose gradient 200,000 g, 16 h Washing step fractions 100,000 g, 90 min | > At early disease stage, an increased number of brain-derived EVs was found, and this was decreased at a late stage > Significant increase in psychosine, myelin proteolipids and myelin basic proteins in brain-derived EVs, which further increased parallel to disease development > Treatment with ceramide production inhibitor GW4869 significantly decreased the number of brain-derived EVs, which accelerated the acquisition of the final disease state | [81] | |
| MLD | In vivo ASA-/- mice (cortex) In vitro Primary Neural Precursor (NP) and glial cells | Centrifugation (4 °C): 300 g, 10 min 2000 g, 10 min 10,000 g, 30 min 100,000 g, 70 min Wash step 100,000 g, 70 min Sucrose gradient 200,000 g, 16 h Washing step fractions 100,000 g, 70 min Filtration (0.22 μm) Centrifugation: 100,000 g, 90 min Washing step 100,000 g, 90 min | > ASA-/- Cells have a defective PDGFRα pathway: Reduced PDGFRα protein levels and failure to phosphorylate AKT > ASA-/- glia, NP and cortex-derived exosomes contain significantly increased PDGFRα levels > Enzyme correction of ASA-/- NPs normalized PDGFRα levels, led to re-localization of the receptor in detergent-resistant membrane domains, increased AKT phosphorylation, normalized the production of oligodendrocytes and reduced exosomal shedding > PDGFRα is secreted in vivo through exosomes during the peak of myelination | [82] |
| Sialidosis (type I and II) | In vitro Neu1-/- mice myofibroblasts Human skin fibroblasts | Centrifugation (4 °C): 300 g, 10 min 2000 g, 10 min 10,000 g, 30 min 100,000 g, 2 h Washing step 100,000 g, 2 h Sucrose gradient 100,000 g, 2.5 h | > In Neu1-/- myofibroblasts, there is excessive lysosomal exocytosis and significantly increased secretion of EVs > Neu1-/- myofibroblasts-derived EVs contain molecules of the TGF-β and WNT signaling pathways Several molecules were present on the outer membranes of EVs through glypicans > Neu1-/- myofibroblasts and fibroblast EVs induce proliferation and migration/invasion in WT cells through profibrotic signals > Neu1-/- myofibroblasts EVs convert normal fibroblasts into myofibroblasts through significantly upregulating genes encoding members of the TGF-β and WNT signaling pathways | [78] |
| Niemann-Pick Disease type C | In vitro NPC1-/- liver cells from BALB/c mice | Centrifugation: 100,000 g, 60 min | > 2-hydroxypropyl-β-cyclodextrin (HPB-CD) treatment stimulates lysosomal exocytosis and EV secretion in a calcium-enhanced manner, which ameliorates endolysosomal cholesterol storage | [83] |
| In vitro Human NPC1-/- skin fibroblasts and CT43 CHO cells; NPC1 siRNA treated oligodendroglia (Oli-neu) | Centrifugation: 3000 g, 10 min 4000 g, 10 min 4000 g, 10 min 10,000 g, 30 min 100,000 g, 60 min Sucrose gradient 200,000 g, 18 h Washing step 100,000 g, 1 h | > NPC1-/- fibroblasts secrete significantly higher amounts of EVs; > NPC1-/- CHO cells secrete significantly higher amounts of EVs containing cholesterol; > Cholesterol enhances the release of flotillin-2-positive EVs | [76] | |
| In vitro NPC1-/- fibroblasts NPC1 KO HeLa cells | Centrifugation (4 °C): 2.000 g, 30 min 10,000 g, 30 min 100,000 g, 90 min | > Treatment of NPC1-/-cells with LBPA precursor phosphatidylglycerol (PG) reduces endolysosomal cholesterol accumulation through increased secretion in EVs containing cholesterol | [77] | |
| Niemann-Pick Disease type B | In vitro Human ASMase-/- B lymphocytes | TEM and Flow Cytometry | > LAMP-3/CD63 accumulation, which points to impairment of endocytic trafficking > Microvesicle/lipid particle release may partially bypass the trafficking blockage caused by lipid accumulation > Rapamycin might regulate autophagy/mitophagy and contribute to the clearance of lipid storage by vesicle secretion and lysosomal exocytosis | [84] |
Therapeutic stimulation of EV release may have the potential as (pre-)treatment for LSDs through the reduction of LSD specific molecule accumulation inside the cell. This could lower the secondary effects on the endolysosomal pathway by ameliorating the accumulation of MVBs. Advantageous effects of autophagy suppression on the secretion of EVs have been shown in Alzheimer’s Disease and PD[85,86]. In these studies, autophagy inhibition by ammonium chloride or bafilomycin A treatment led to the release of EVs containing the accumulated molecules alpha-synuclein and amyloid precursor proteins[85,86]. A similar effect was observed in Pompe Disease, where ATG7 was inactivated in the mouse muscles. The autophagy suppression alone led to a 50%-60% reduction of the accumulating glycogen, and after ERT delivery, this was restored to normal levels[87]. Unfortunately, the effects on EV secretion were not tested in this study, and it appeared that the accumulating molecule was digested in the cytosol, which improved the cellular phenotype. Contradictory to these results, other studies on Pompe Disease showed improvements in myotubes and muscle tissue after induction of autophagy through TFEB overexpression[88,89]. Moreover, induction of autophagy with rapamycin has been shown to lead to higher secretion of lipid particles and CD63+ vesicles in Niemann-Pick type B. This data is in line with studies that predict a way in which autophagy-inducing conditions, mainly rapamycin treatment, have a specific role in unconventional secretion[90]. It must be noted that in LSDs, there is no problem with the formation of autophagosomes. Autophagy problems are secondary to dysfunctional lysosomes that create a blockage in the autophagic flux and alter this pathway. Therefore, it must carefully be evaluated to what degree each mutation affects the endolysosomal pathway in all LSDs separately. Alongside the effect of TFEB on autophagy, it can increase lysosomal biogenesis and lysosomal exocytosis. Lysosomal exocytosis could be an alternative route to induce cellular clearance and has been implicated as a mechanism used by MLD and mucolipidosis type I cells[91,92]. Lysosomal exocytosis can also be induced by drugs such as 2-hydroxypropyl-β-cyclodextrin (HPB-CD). HPB-CD is used as excipient to facilitate the transport of molecules across membranes as well as treatment to manipulate cellular cholesterol levels. The drug is approved by the FDA and was already used to alleviate cholesterol accumulation in Niemann Pick Type C patients[93]. Treatment with HPB-CD can induce calcium-dependent lysosomal exocytosis and EV release[83].
While improving the cellular state sounds promising, the secretion of EVs by LSD cells could have negative effects on neighboring cells or tissues. Their cargo could contribute to the progression of the pathological conditions over time, considering that EVs from LSD cells can contain the accumulated metabolites[76,94]. This could especially have a major impact on neuronal tissue and avascular connective tissue. Both tissue types are hard to reach and treat due to the lack of blood supply or barriers, which results in the progression of symptoms after the first line of treatment[15]. Increased secretion of the accumulating metabolites could further alter the carefully regulated composition and spatial orientation of the extracellular matrix (ECM). Alterations in the ECM are often seen in LSDs and mostly in MPS, where the primary storage material is a component of the ECM. The specific alterations depend on the type of LSD and may include changes in the size, expression, and arrangement of both fibrous elements and proteoglycans. Furthermore, alterations in the levels of hyaluronic acid (HA) and matrix metalloproteinases (MMPs) are often reported. The ECM modifications in LSDSs can have effects on multiple signaling pathways, cell-matrix and cell-cell interactions[95]. The contributions of EVs to these processes were shown by Van de Vlekkert et al., who demonstrated that Neu1−/− myofibroblasts derived-EVs were able to convert normal fibroblasts into myofibroblasts propagating fibrosis[78]. This effect was obtained through upregulation of genes in the TGF-β and WNT signaling pathways[78]. This suggests that Neu1−/− myofibroblasts derived-EVs can contribute to and/or cause progression of fibrosis. Furthermore, in Krabbe Disease, accumulation of psychosine results in more rigid myelin fluidity and demyelination which occurs through the shedding of myelin by microvesicles[79]. This deregulates cell signaling and may introduce focal weak points. In a healthy situation, myelin covers and protects nerve cells to ensure rapid transmission of nerve signals. Consequently, demyelination interrupts signaling of the central and peripheral nervous systems, which contributes to the neurological symptoms seen in Krabbe patients[96]. In additional Sphingolipidoses Niemann-Pick type A and C[97,98], changes in the membrane organization and decreased fluidity have been reported. This decrease in membrane fluidity is linked to altered activity of membrane-bound enzymes, carrier-mediated transport, and receptor binding at the cell membrane[98]. Whether this influences EVs release in these LSDs remains to be determined. To study EV release in Fabry Disease, Song et al. used CRISPR/Cas editing to create GLA-null human embryonic stem cells (hESCs)[68]. This model for hypertrophic cardiomyopathy showed impaired autophagic flux and protein turnover combined with increased EV release[68]. These studies show the contribution of EVs to the clinical manifestations of LSDs, such as fibrosis, delamination, and hypertrophic cardiomyopathy. More research is needed to investigate the role of the secondary cascade following lysosomal dysfunction in individual LSDs. This will greatly improve our understanding of its pathophysiology inside the cell and its contribution to disease progression. In addition, this could contribute to more targeted and more efficient therapy development. For now, these studies on EVs have demonstrated the potential of vesicles as biomarkers for these symptoms.
THE POTENTIAL OF EVs AS BIOMARKERS FOR LSDs IN DIAGNOSTICS AND DISEASE MONITORING
EVs are able to reflect the metabolic status of the parent cell through alterations in their cargo, including RNAs, lipids, sugars and proteins[37]. They can be isolated from patient material, such as blood, urine, saliva, tear fluid, cerebrospinal fluid, and amniotic liquid[99-101]. Moreover, the concentration of EVs in fluids seems to be disease-state dependent as well[60]. Therefore, EVs are considered a novel and promising tool in diagnostics and their role as biomarkers has been well studied in multiple diseases with similarities to LSDs, including Alzheimer’s Disease[102] and PD[103]. However, their potential as biomarkers in LSDs has yet to be explored.
Since most LSDs are elusive at birth, the diagnostic process of LSDs often relies on suspicion of the physician based on clinical manifestations. However, early diagnosis of LSDs is of utmost importance to start early treatment and prevent irreversible damage to all organ systems involved[104]. Current diagnostics of LSDs rely on genetic testing, analyses of undegraded metabolites, and enzymatic activity with dry blood spots (DBSs). Testing on undegraded material is mostly performed on plasma and urine samples; however, this is only available for a limited number of LSDs and lacks specificity. Therefore, confirmation mostly relies on enzymatic testing with DBSs in high-throughput platforms, which is only possible for enzyme deficiencies[105,106]. Until now, research on the diagnostic potential of EVs in LSDs is limited. Nevertheless, it has been shown that EVs isolated from Niemann-Pick type C cells contain the accumulated metabolite cholesterol and MLD-EVs contain the accumulated sulfatides[76,94]. This might be the case for EVs in other LSDs as well, as the content of EVs reflects the contents and state of the EV-producing cells. Thus, collection of EVs from different types of body fluids could be a valuable source of biomarkers. Their prediction efficiency as biomarkers could be dependent on the location of specific LSD symptoms. For instance, tear fluid has been shown to contain approximately 100-fold higher EV concentrations than plasma[101]. Since ocular symptoms such as corneal clouding are often present in LSDs, EVs isolated from tear fluid could be a suitable candidate for biomarker analysis. In other ocular diseases, it has been shown that isolated EVs contain a distinctive protein pattern compared to healthy controls[107]. Furthermore, enzyme activity studies can be performed on tear samples as previously shown for MPS I[108]. These findings warrant EVs to be studied as a potentially valuable biomarker source in diagnostics, monitoring of disease progression and analysis of treatment efficiency in the future.
As stated before, early indication of LSDs is extremely important to start effective treatment and prevent the development of organ damage. Amniotic fluid could be used for the detection of LSDs. However, most LSDs are typically not evaluated for in the initial standard-of-care workup when babies are born. In several states in the US, parent advocacy has led to the expansion of NBS programs that now include newborn screening in dried blood spots (DBS) for Pompe, Krabbe, Gaucher, Fabry, MPS-I, MPS-II, and Niemann-Pick A/B or a subset[109], while in the Netherlands the screening only includes MPS I[110]. Firstly, more LSDs and other metabolic disorders could be included in the standard workup[111]. Secondly, when parents choose to have an amniocentesis, co-collection of an amniotic fluid sample could be performed. Isolation of amniotic EVs from these samples could potentially improve early diagnosis. However, as performing an amniocentesis has several risks, collection of EVs from amniotic fluid could be considered alongside scheduled amniocentesis. Keller et al. showed that it is possible to separate EVs derived from the fetus and the mother and that these EVs partly represent the renal system of the fetus[112]. Even early in development, these EVs could already carry the undegraded material in case of a LSD, as this can normally be detected in urine samples of newborns[105]. Using EV-based biomarkers in the diagnostic process may hold the potential to improve the speed of diagnosis and clinical risk assessment of LSDs early in development. In general, the use of EVs could increase the sensitivity of biochemical assays by increasing the concentration of test material. If successful, this could improve both maternal and fetal patient management[113].
Until now, biomarker research in LSDs is limited to the enzyme deficiencies Fabry, Gaucher and MLD. Table 3 shows an overview of all literature related to EVs and their role as biomarkers in these LSDs. EVs could play a role in real-time analysis of disease progression, which could improve personalized treatment protocols. Following this idea, Levstek et al. studied urine-derived EVs (uEVs) to evaluate their potential in the prediction of nephropathy progression in Fabry Disease[114]. In almost all Fabry patients, the kidneys are affected. In this study, it was shown that uEVs from Fabry patients with nephropathy contained increased expression of miR-29a-3p and miR-200a-3p. On the other hand, patients who were not affected by renal dysfunction had higher expression levels of miR-30b-5p, which plays a protective role in podocyte injury. This shows that screening of uEVs in these patients could help define the presence and status of nephropathy[114]. For patients with some residual enzyme activity, this method could be used to assess when to start ERT, while for patients with little or no residual enzyme activity, this could mark the start of dialysis[115]. Gaucher Disease includes a major genetic risk factor for the development of PD in these patients. Tatiana et al. performed proteomic profiling of EV proteins from blood plasma of Gaucher patients to find any association with PD[116]. While they showed increased EV size in patient material, proteomics did not reveal any PD-associated proteins[116]. Additional EV cargo, such as lipids, sugars and RNAs, were not tested in this study.
Extracellular vesicles as biomarkers for LSDs
| LSD | Study design | Patient material | EV cargo (biomarker) | Isolation method | Major results | Reference |
| Fabry Disease | Patient material | Urine | Negative: miR-29a-3p and miR-200a-3p Positive:miR-30b-5p | Centrifugation: 2000 g, 15 min Storage -80 °C Mix urine with PBS and EDTA AMICON Ultra-15 Spin-filter 4000 g, 10 min SEC (70 nm column) AMICON Ultra-4 Spin-Filter 4000 g, 6 min at 4 °C | > uEVs contain increased expression of miR-29a-3p and miR-200a-3p over time in patients with nephrology > Over 10 years, uEVs of patients without renal dysfunction contained increased levels of miR-30b-5p, which has a protective role in podocyte injury | [114] |
| Patient material In vitro HUVEC | Plasma | miR-126-3p | Plasma EVs Centrifugation: 1500 g, 10 min 3000 g, 30 min 10,000 g, 30 min 100,000 g, 1 h 45 min at 4 °C HUVEC EVs Centrifugation: 300 g, 10 min 2000 g, 20 min 10,000 g, 30 min 110,000 g, 70 min Washing step 110,000 g, 70 min | > Circulating miR-126 levels physiologically increase with age > sEVs of Fabry Disease patients contain higher miR-126-3p levels > Glycosphingolipid accumulation induces premature senescence in HUVECs and increases miR-126-3p levels > Fabry Disease may aggravate the normal aging process | [117] | |
| MLD | In vivo MLD mice (ARSA-/-) | HIn vivo emi sagittal brains | Blade dissociation 100 μm mesh filtration 40 μm mesh filtration Centrifugation: 300 g, 10 min 2000 g, 20 min 10,000 g, 30 min 10,000 g, 30 min 100,000 g, 90 min Sucrose gradient 200,000 g, 16 h 100,000 g, 90 min | > Brain derived-EV concentration increased in parallel with age > MLD brain-EVs are significantly larger at 3 months compared to day 30 or 6 months > There is loading of accumulating sulfatides into MLD-EVs and alterations in the loading of different ceramides > Progressive decrease of FAs and FAHFAs in MLD-EVs | [94] | |
| Gaucher Disease | Patient material | Plasma | Increased size | Centrifugation: 2000 g, 30 min 16.000 g, 30 min 110,000 g, 2 h Washing step Gentle swaying for 1h at 4 °C Filtered (0.45 μm) | > EVs of Gaucher Disease patients are increased in size and have altered morphology > Proteomic profiling of exosomal proteins did not reveal any proteins associated with PD pathogenesis | [116] |
| Patient material | Serum | Increased number of MPs | Centrifugation (20 °C): 1550 g, 20 min 18.800 g, 30 min Washing step 18.800 g, 30 min | > Higher levels of MPs from different blood cells in patient samples both before and after ERT compared to controls > After 1 year of ERT, the total MPs in GD were significantly decreased | [118] |
In addition to protein and miRNA content, the lipid composition of EVs could be involved in the reflection of disease pathology. Hence, Pergande et al. performed an untargeted lipidomic analysis of brain-derived EVs isolated from MLD mice[94]. In the sphingolipidosis MLD, deficiency of the aryl-sulfatase A (ARSA) enzyme leads to accumulation of sulfatides, including multiple sulfated glycolipids[94]. The accumulation of sulfatides causes severe neurological symptoms in MLD patients. Sulfatides have been found in EVs before, for instance, in EVs isolated from plasma and oligodendrocytes[119,120]. Pergande et al. showed that MLD-EVs contained numerous altered lipid species from a broad range of lipid classes[94]. This included sulfatides and their ceramide precursors as well as fatty acids (FAs), fatty acid esters of hydroxyl fatty acids (FAHFAs) and acylglycerol lipids. The sulfatides include mostly 18-carbon fatty acid chains, which predominantly accumulate in MLD mice. Furthermore, these lipid alterations in EVs were found to be age-dependent and are proposed as relevant candidates for MLD biomarkers[94].
Further research into specific LSD-related biomarkers could improve our understanding of disease pathology as well as improve patient care. Moreover, unraveling the tissue origin of EVs secreted in different body fluids through specific tissue biomarkers could improve our understanding of disease location and progression and thus improve treatment. We suspect that the use of algorithms in EV research will progress in the upcoming years and will be of great value to analyzing EV data from body fluids in relation to disease[51,121-123].
IMPROVEMENT OF THERAPEUTIC DELIVERY WITH EXTRACELLULAR VESICLES AS DRUG DELIVERY VEHICLES
EVs are biocompatible and have intrinsic tissue-penetrating abilities[124]. In addition, EVs show low immunogenicity and are stable in circulation due to their negatively charged surface and their ability to avoid the mononuclear phagocytic system upon modification[62]. These features qualify EVs as potential new therapeutic drug delivery vehicles. The ability of EVs to cross the BBB could open doors to the development of novel treatment and delivery strategies for LSDs with neurological deficits[38]. Furthermore, it makes EVs a more desirable delivery tool over existing nanotechnologies such as liposomes, synthetic polymers and gold nanoparticles[125,126]. EVs naturally contain lysosomal components including heparan-alpha-glucosaminide N-acetyltransferase (HGSNAT)[50], beta-glucocerebrosidase (GBA), N-acetylgalactosamine-6-sulfate sulfatase (GALNS)[127], cystinosis (CTNS)[128], deficient in MPS III, Gaucher, MPS IVA, and Cystinosin respectively. Considering the interplay between the endolysosomal pathway and EV secretion, there could be a broader range of natively loaded LSD deficient proteins into EVs[128], making them an interesting vehicle for ERT.
EV uptake into recipient cells can take place through a variety of different pathways. EV internalization predominantly occurs through endocytosis, which includes macropinocytosis, receptor-mediated, clathrin-dependent and clathrin-independent mechanisms[129]. In addition, EVs could also enter the cells through phagocytosis. Additionally, EVs may also exert phenotypical effects on target cells without entry through direct receptor-ligand interactions[124,130]. Most of the internalization pathways lead EVs to the endolysosomal pathway. Numerous studies have shown strong colocalization of EVs with lysosomes, reaching up to ~50%-60% in fibroblasts[130]. For most therapies, this will be a disadvantage when (intraluminal) EV cargo needs to be delivered to the cytosol of nucleus. In that case, EVs need to escape the endosome to avoid degradation in the lysosome. It is largely unknown how EVs can naturally induce endosomal escape; however, there is some evidence that fusion between the membranes of EVs and the late endosomal membranes plays a role[124], potentially under the influence of acidification in the endolysosomal environment[131]. Nevertheless, EVs being trafficked towards the lysosome could be of substantial benefit for the delivery of therapeutics in the treatment of LSDs. However, whether the same distribution pattern towards the dysfunctional lysosomes in LSD cells will be seen remains to be determined.
Already a decade ago, Coulson-Thomas et al. showed that transplantation of human umbilical mesenchymal stem cells improves the corneal defects of MPS-7 mice[132]. After injections of these MSCs, improvements were hypothesized to be mediated through MSC-derived EVs[132]. At the time, EVs were gaining more recognition for their potential therapeutic abilities[133]. To date, several studies have shown that overexpression of lysosomal enzymes in cells leads to passive loading into EVs, including the deficient enzymes of Fabry[128], MPS IIIA[128], MPS IVA[127], and Batten Disease[134]. An overview of all studies regarding EVs in the treatment of LSDs can be found in Table 4.
Extracellular vesicles as therapeutic carriers for LSD treatment
| LSD | Study design | EV cargo | EV concentration | Isolation method | Major results | Reference |
| Fabry Disease | EVs from stable GLA expressing CHO DG44 cells In vitro HEK293T and NAEC cells In vivo GLA-/- mice | GLA | In vitro 2.5 μg EVs/ml (c.a. 125 ng GLA/ml) In vivo 1 mg/kg GLA | Centrifugation (4 °C): 3900 rpm, 15 min (free enzyme) 300 g, 10 min 2000 g, 10 min 10,000 g, 20 min VIVAspin 30,000 KDa 7.000 g, 10 min Precipitation o/n with ‘Total Exosome Isolation Reagent’ 16.000 g, 1 h | > Significant amounts of GLA in EV lysates compared to whole cell lysates > Predominant contribution of clathrin-mediated endocytosis and macropynocytosis in EV uptake > EVs protect GLA and increase its enzymatic activity upon delivery compared to commercial GLA and agalsidase alfa used in ERT > EVs were well tolerated in vivo and distributed among all main organs, including the brain after i.a. administration | [128] |
| MPS IIIA | Transient SGSH expressing HEK293 cells In vitro HEK293T and NAEC cells | SGSH | 2.5 μg EVs/ml | Centrifugation (4 °C): 3900 rpm, 15 min (free enzyme) 300 g, 10 min 2000 g, 10 min 10,000 g, 20 min VIVAspin 30,000 KDa 7000 g, 10 min Precipitation o/n with ‘Total Exosome Isolation Reagent’ 16.000 g, 1 h | > Significant amounts of SGSH in EV lysates compared to whole cell lysates > EVs restore lysosomal function much more efficiently than the recombinant enzyme in clinical use > In vivo, EVs were well tolerated and distributed among all main organs, including the brain | [128] |
| MPS IVA | hUMSC-derived EVs In vitro MPS IVA deficient patient fibroblasts | GALNS | Co-cultures | Conditioned medium 3000 rpm, 15-20 min at 4 °C Centrifugation: 300 g, 10 min 10,000 g, 30 min 0.22-μm filter 100,000 g, 90 min | > UMSC-EVs containing functional GALNS enzyme and can deliver it to CALNS deficient cells; > Mannose-6-phosphate inhibition did not affect enzyme delivery after EV treatment; > EVs are a function and novel technique for reducing GAG accumulation in cells of avascular tissues | [127] |
| Cystinosis | amMSCs and bmMSCs In vitro Patient derived CTNS-/- skin fibroblast and ciPTEC | CTNS | Co-cultures, 1-2 mg microvesicles/mg fibroblast protein | Centrifugation (4 °C): 300 g, 5 min 100,000 g, 2 h Washing 100,000 g, 1 h Washing 100,000 g, 1 h | > Amniotic fluid or bone marrow derived hMSCs, reduce pathologic cystine accumulation in co-culture with patient CTNS-/- fibroblasts or proximal tubular cells; > MSC derived EVs contain wildtype cystinosin protein and CTNS mRNA | [135] |
| EVs derived from Spodoptera frugiperda (Sf9) cells infected with Baculovirus In vitro CTNS-/- fibroblast | CTNS | - | 0.2 μm filter Dialysis to dilution of 1/2500, or 2 (cut-off 3.5 kDa) Centrifugation: 140,000 g, 3 h followed filter-sterilization | > Vesicle addition to CTNS-/- fibroblast leads to 57% cystine depletion > LC-MS/MS analysis of the vesicles shows three cystinosin peptides with 7.4% coverage of the human cystinosin sequence > Quantitative tandem mass spectrometry showed 0.1 and 1.2 pmol cystinosin per mg of vesicle protein > Both western blot and RT-PCR confirmed the presence of cystinosin in these vesicles | [136] | |
| EVs derived from Spodoptera frugiperda (Sf9) cells infected with Baculovirus containing the hCTNS-GFP In vitro CTNS-/- fibroblast Rabbit globes | CTNS | Gas-permeable stoppers containing 50 ml medium with or without the 1011/ml EVs | Freeze-thaw snap freezing medium Thawed Dialyzed into Ham’s F12, 48 h at 1:2500 dilution 0.22 μ filter Stored at 4 °C | > Baculovirus infected Spodoptera frugiperda cells (Sf9) spontaneously produce EVs that contain cystinosin > EVs are detected in CTNS-/- fibroblast with perinuclear and cytoplasmic distribution after treatment; > Upon ex vivo ocular EV delivery, GFP signal is detected for 48 h | [137] | |
| CLN2 | TPPI-plasmid DNA transfected peritoneal macrophages or sonication and permeabilization of EVs with saponin to load TPPI In vitro CLN2-/- skin fibroblasts In vivo Intraperitoneal EV administration in late-infantile neuronal ceroid lipofuscinosis (LINCL) mice | TPPI | In vitro 1010/mL In vivo Intraperitoneal administration Bioimaging: 2 × 1011 in 200 μL saline Brain accumulation: 1 × 1010 in 100 μL Therapeutic efficiency: 4.3 × 1012 in 150 μL | Centrifugation: 300 g, 10 min 1000 g, 20 min 10,000 g, 30 min 0.2 μm syringe filter 100,000 g, 4 h Washing 120,000 g, 70 min Washing 120,000 g, 70 min Sonification or permeabilization | > Both transfection with TPPI plasmid and sonification/permeabilization lead to proficient incorporation of functional TPP1 into macrophage-EVs (EV-TPP1); > EVs significantly increase stability of TPP1 against protease degradation and provide efficient TPP1 delivery to CLN2-/- fibroblasts; > Around 70% of the EV-TPP1 is delivered to lysosomes; > Intraperitoneal administration of EV-TPP1 leads to accumulation in the brain of LINCL mice, which increased lifespan | [134] |
| EVs derived from mice bone marrow-derived macrophages In vitro CLN2-/- primary cortical neurons In vivo Intrathecal and intranasal routes CLN2-/- LINCL mice | TPPI | In vitro 5 × 1011 EVs In vivo i.v. 6 × 1011 in 200 µL i.p. 6 × 1011 in 200 µL i.t. 1.5 × 1011 in 50 µL i.n. 6 × 1011 in 20 µL | Centrifugation: 300 g, 10 min 1000 g, 20 min 10,000 g, 30 min 0.2 μm syringe filter 100,000 g, 4 h Washing 120,000 g, 70 min Washing 120,000 g, 70 min Sonification | > EV-TPP1 treatment though i.t. and i.n. routes lead to accumulation in the brain, decreased neuroinflammation and neurodegeneration and reduced aggregation in CLN2-/- mice; > EV-TPP1 treatment though i.v. and i.p, administration leads delivery to the liver, spleen, kidney, and lungs; > Combined i.t. and i.p. injection significantly prolonged lifespan in CLN2-/- mice | [138] |
While EVs improve drug stability and circulation time[139], they can be further optimized to improve EV uptake and biodistribution and obtain targeted delivery. All cells release EVs, but the number, composition, functionality and physicochemical characteristics vary between cell sources[62]. For instance, MSC-derived EVs have immunomodulatory and anti-inflammatory effects. Recently, unmodified MSC-EVs were used to treat osteoarthritis and resulted in amelioration of disease progression through induced polarization of M2-type macrophages among other effects[140]. These positive effects on disease related to connective tissue could be beneficial to treat residual disease and target the connective tissue in LSD patients. Furthermore, the native characteristics of MSC-EVs could be further exploited through engineering. This might lead to delivery of the loaded protein while also promoting tissue repair and regeneration within the same treatment. Moreover, EV functionality is dependent on the target as well, considering that some EVs are better taken up by one cell type compared to another[36]. Therefore, therapy specific selection of the optimal cell source, cell cultivation procedure, EV extraction and purification methods, and storage might be needed.
Recently, Seras-Franzoso et al. showed that stabile GLA expressing cell lines were able to produce EVs that protected the enzymes from proteases, and subsequently improved enzymatic delivery directly compared to soluble GLA and clinically approved ERT agalsidase alfa[128]. In addition, analysis of the GLA biodistribution showed that EVs were able to deliver significantly more active enzymes to the kidneys than non‐encapsulated enzymes. It must be noted that agalsidase alfa used in ERT is governed by the presence of the mannose-6-receptor to increase uptake. This receptor is highly expressed in the liver and therefore might interfere with the biodistribution of non-encapsulated enzymes [141]. Furthermore, they showed the uptake of fluorescent labelled EV-GLA in the brain parenchyma with confocal microscopy an hour post-intra‐arterially (i.a.) administration through cannulation of the external carotid artery. This delivery across the BBB could not be detected after intravenous administration through the tail vein[128]. Likewise, Haney et al. showed the loading of tripeptidyl peptidase-1 (TPP1) into EVs, which greatly improved enzyme stability[134,138]. After showing functional delivery of the TPP1 in EV treated fibroblast, the authors examined intraperitoneal administration of EVs in late-infantile neuronal ceroid lipofuscinosis (LINCL) mice to study EV distribution towards the brain. This type of administration led to accumulation of the enzyme in the brain. However, a clear distribution towards peripheral organs was also seen, including the liver, spleen, kidneys and lungs[134]. Therefore, they examined different EV administration routes in mice in another study to see the effect on crossing the BBB. Both intranasal and intrathecal administration resulted in accumulation of TPP1 in the brain and spinal cord, which reduced aggregation of lysosomal storage material, neurodegeneration and neuroinflammation. Importantly, these administration routes showed lower distribution of the EVs towards the peripheral organs compared to intraperitoneal and intravenous administration[138]. The loading of enzymes into EVs and their distribution towards the central nervous system (CNS) shows the potential of EVs as therapeutic carriers to treat the CNS. Thus, EVs could be used to load and protect ERT to improve biodistribution and lower antibody generation in patients.
ENGINEERING OF EXTRACELLULAR VESICLES FOR THERAPEUTIC TREATMENT OF LSDs
While native EVs have multiple features that qualify them as promising drug delivery vehicle, endogenous cargo loading, circulation time and tissue targeting are often inadequate. Biodistribution studies often show accumulation of EVs in the liver, spleen, gastrointestinal tract and lungs. However, the specific pattern of distribution seems to depend on the cell type the EVs are derived from, the route of administration[142] as well as the injected dose[64]. Several studies have demonstrated a role for integrins in tissue targeting[143,144]. Integrins are enriched in EVs through their association with tetraspanins[145]. For instance, integrin expression patterns in tumor-derived EVs play a role in the uptake of EVs by organ-specific cells and creation of a pre-metastatic niche[143]. The use of fluorescent labelling and tracking techniques are valuable tools to study and optimize the distribution pattern of (engineered) EVs. For an overview of the currently used labelling and tracking techniques in the EV field, we refer to the review of Kooijmans et al.[129]. To further optimize performance, EVs can be modified and engineered. To date, studies on EV engineering for LSD treatment have solely focused on actively increasing therapeutic cargo loading. These modifications can be obtained through a variety of different exogenous and endogenous methods. In exogenous techniques, EVs are therapeutically loaded post-isolation by simple incubation, electroporation, sonication, extrusion, and freeze-thawing. These techniques have variable degrees of success and can lead to EV or cargo aggregation[146,147]. Uniquely, EVs can also be endogenously altered by biologically engineering the cells that produce them. This strategy can equip EVs with new moieties while preserving their membrane integrity, which is an advantage over exogenous loading techniques[123].
Therapeutic protein loading into EVs is often achieved through parental cell engineering with genes that encode for the therapeutic protein fused to a membrane protein enriched in EVs or a transmembrane domain. These loading strategies may include the use of EV enriched proteins such as tetraspanins (CD9, CD63 and CD81), PTGFRN, and BASP1[148] or transmembrane domains such as the N-terminal fragment of syntenin[149]. With this strategy, the therapeutic protein will naturally be incorporated into EVs during biogenesis. This approach allows for both internal and external display of the therapeutic protein[150]. Successful protein loading and delivery, both in vitro and in vivo, has been shown in multiple studies[150-152]. This shows the potential for active enzyme loading and accumulation in EVs to increase functional delivery. When designing active enzyme loading strategies, it must be considered that lysosomal enzymes often require post-translational modification. These modifications need to take place before loading enzymes into EVs to eventually lead to functional delivery upon EV treatment[153,154].
To date, the only LSD for which active enzyme loading into EVs has been studied is Gaucher Disease. An overview of these studies can be found in Table 5. In patients with Gaucher Disease, the GBA enzyme is deficient, which leads to symptoms that include neurological complications. Do et al. showed active GBA loading into EVs with the use of vesicular stomatitis virus glycoprotein (VSV-G)[153]. In addition to loading and potential uptake through the VLDL receptor on the surface of recipient cells, VSV-G can also induce endosomal escape[90]. It should be noted that induced endosomal escape could potentially counteract the goal of this EV treatment by enabling the EV cargo escape their route to the lysosome. Despite that, Do et al. show significant colocalization of the EVs throughout uptake with the endosome, late-endosome and lysosome in an in vitro HEK293 cell model[153]. However, it is possible that delivery of GBA to these cellular structures would even further increase with the use of a loading strategy that does not additionally induce endosomal escape. Flow cytometry analysis showed a 73%-75% uptake rate based on mean fluorescent intensity of the co-loaded GFP[153]. Although this study focused on in vitro cell culture models, it underlines the potential of active loading strategies into EVs to deliver deficient enzymes as next-generation ERT. In the future, such therapies may lower antibody generation to ERT in patients and deliver functional enzymes to target organs including the central nervous system (CNS), which is severely affected in multiple LSDs.
Engineered extracellular vesicles to treat LSDs
| LSD | Study design | EV cargo | Readout | EV concentration | Isolation method | Major results | Reference | |
| Gaucher Disease | EVs from HEK293T cells Active loading VSVG-GFP-GBA | GBA | In vitro HEK293T, U7 and HepG2 | 0.3 µg/μl | Centrifugation: 1500 g, 10 min 0.2 µm syringe filter Mixed with ExoQuick-TC solution (1:4) o/n at 4 °C 3000 g, 90 min at 4 °C | > HEK cells transfected with VSVG-GFP-GBA constructs secrete EVs that contain significantly more active GBA compared to control EVs; > The uptake of green, fluorescent dye labelled GBA-EVs was 73%-75%; > Upon treatment, GBA loaded EVs co-localize with lysosomes and lead to the delivery of functional GBA (40%-45%) | [153] | |
| CLN2 | TPPI-plasmid DNA transfected peritoneal macrophages or sonication and permeabilization of EVs with saponin to load TPPI In vitro CLN2-/- skin fibroblasts In vivo Intraperitoneal EV administration in late-infantile neuronal ceroid lipofuscinosis (LINCL) mice | TPPI | In vitro IC21 cells, neuronal PC12 cells and Human skin CLN2-/- fibroblasts In vivo LICL mice | In vitro 1010/mL In vivo Intraperitoneal administration Bioimaging: 2 × 1011 in 200 μL saline Brain accumulation: 1 × 1010 in 100 μL Therapeutic efficiency: 4.3 × 1012 in 150 μL | Centrifugation: 300 g, 10 min 1000 g, 20 min 10,000 g, 30 min 0.2 μm syringe filter 100,000 g, 4 h Washing 120.000 g, 70 min Washing 120,000 g, 70 min Sonification or permeabilization | > Both transfection with TPPI plasmid and sonification/permeabilization lead to proficient incorporation of functional TPP1 into macrophage-EVs (EV-TPP1) > EVs significantly increase stability of TPP1 against protease degradation and provide efficient TPP1 delivery to CLN2-/- fibroblasts > Around 70% of the EV-TPP1 is delivered to lysosomes > Intraperitoneal administration of EV-TPP1 leads to accumulation in the brain of LINCL mice, which increased lifespan | [134] | |
| EVs derived from mice bone marrow-derived macrophages In vitro CLN2-/- primary cortical neurons In vivo Intrathecal and intranasal routes CLN2-/- LINCL mice | TPPI | In vitro Primary bone murine marrow-derived macrophages, cortical neurons and IC21 cells In vivo LICL mice | In vitro 5 × 1011 EVs In vivo i.v. 6 × 1011 in 200 µL i.p. 6 × 1011 in 200 µL i.t. 1.5 × 1011 in 50 µL i.n. 6 × 1011 in 20 µL | Centrifugation: 300 g, 10 min 1000 g, 20 min 10,000 g, 30 min 0.2 μm syringe filter 100,000 g, 4 h Washing 120,000 g, 70 min Washing 120,000 g, 70 min Sonification | > EV-TPP1 treatment though i.t. and i.n. routes lead to accumulation in the brain, decreased neuroinflammation and neurodegeneration and reduced aggregation in CLN2-/- mice > EV-TPP1 treatment though i.v. and i.p, administration leads delivery to the liver, spleen, kidney, and lungs. > Combined i.t. and i.p. injection significantly prolonged lifespan in CLN2-/- mice | [138] | ||
THE USE OF EVs IN FUTURE THERAPEUTIC TREATMENT OF LSDs
Engineered EVs have already been explored for multiple purposes, including regenerative therapy[155], immune modulation[156], delivery of small molecular drugs[157], vaccines[158], therapeutic proteins, and nucleic acids[159]. For instance, cardiac progenitor cell-derived EVs loaded with miR-322 outperformed their unloaded controls in enhancing angiogenesis in mice after ischemic injury[155]. Furthermore, numerous small molecular drugs have been successfully incorporated into EVs, including curcumin. The loading of curcumin into EVs enhanced its delivery and anti-inflammatory activity[156]. Drug loading into EVs has been shown to improve accumulation in target cells and enhance drug stability and circulation time[139]. Currently, multiple groups are exploring the potential of employing EV engineering for the development of therapeutic treatments. The variety and possibilities of this technique could greatly improve therapeutic options for LSDs in the future.
Alongside the mentioned positive effects of using EVs to deliver ERT, delivery of other forms of LSD treatment might also benefit from this camouflage strategy. This may include SRT and PCT, which use small drugs to inhibit the production of the accumulating substrate and stabilize native structures of mutated enzymes, respectively[26,27]. Loading both SRT and CPT into EVs might improve their distribution and potential delivery to the CNS. Furthermore, newly discovered molecular drugs might benefit from being loaded into EVs as well by increased loading efficiency, protection, improved circulation and crossing of biological membranes. As discussed before, in most LSDs, autophagy is altered and affected cells might reduce their accumulation through secretion in the hope of maintaining homeostasis. Curcumin treatment has been shown to alleviate the phenotype of Niemann-Pick type C through the elevation of cytosolic calcium levels[160]. Additionally, it has been shown that curcumin promotes EV section in cells that represent Niemann-Pick type C, inducing cholesterol shuttling out of the cell[161]. Loading and delivery of curcumin via EVs may be a therapeutic approach to treat Niemann-Pick type C.
In addition to enzyme deficiencies, there are other subgroups of LSDs that include deficiencies in integral membrane proteins, transporters and proteins involved in the post-translational modification, trafficking, or regulation. For these subgroups, there are limited treatment options available. Gene therapy was evaluated to treat Niemann-Pick type C in which patients are missing or have defective NPC1 transporters. Currently, Evox Therapeutics filed a patent to facilitate the loading of NPC1 fused to syntenin/CD63 into EVs (Patent number: WO2019/092287 AI). The use of EVs to transport NPC1 will stabilize the integral membrane protein through the presence of lipid molecules[162]. This strategy of EV engineering could potentially be a step forward in designing treatment strategies for currently untreatable LSDs.
Current challenges of EVs that must be addressed to improve their therapeutic efficacy include their rapid clearance and limited targeted delivery. Possible solutions may involve adaptation of administration routes depending on the target tissue. Local administration might be preferred for connective tissues with limited blood supply, whereas intranasal or intrathecal administration might be beneficial to target the CNS[138]. Moreover, modification of the EV surface may improve EV circulation and tissue targeting[63,163,164]. Kamerkar et al. have reported that the presence of CD47 on EV surface inhibited EV uptake and clearance by macrophages and monocytes, which may lead to increased cargo delivery[165]. However, considering that LSDs are monogenetic disorders, the monocyte-macrophage system is affected in multiple LSDs as well[166]. Therefore, the uptake of drug-loaded EVs by these immune cells could also be beneficial. Several bioengineering strategies have been developed to increase the affinity of EVs for specific recipient cells. These approaches include receptor-ligand, enzymatic, and antigen-antibody or a combination of these methods. For instance, EVs containing a LAMP2b-designed ankyrin repeat protein (DARPin) G3 fusion protein are able to specifically target HER-2 positive breast cancers through receptor-ligand interaction. DARPins are a class of synthetic peptides that can bind biological receptors with high specificity and binding affinity and specificity[167]. An adaptation of the EV membrane that both improved circulation time and cell specificity was shown by the addition of epidermal growth factor receptor (EGFR) nanobodies conjugated to phospholipid (DMPE)-polyethylene glycol (PEG). The shielding properties of PEG compromised cell binding and increased detectability of EVs in the plasma from less than 10 min to over 60 min post-injection. At the same time, the EGFR nanobodies directed the EVs towards tumor cells overexpressing the EGFR[63]. Surface modification can also be used to evade phagocytosis by the mononuclear phagocyte system (MPS). These strategies will increase circulation time which will improve the chance of EV uptake by the specific recipient cells and can be used to increase EV accumulation and cargo delivery in targeted tissues[63]. Alongside biological engineering, EVs can externally be altered through chemical or synthetic engineering by using click chemistry or fusion with nanotechnologies[62,129]. All the different engineering strategies make it possible to optimize EVs to potentially target any tissue and disease.
EV-MEDIATED GENE EDITING: THE POTENTIAL ROUTE TO A PERMANENT CURE FOR LSDs
LSDs are genetic disorders and recent studies have been exploring the possibilities of gene editing to correct their mutations[168]. CRISPR/Cas technology possesses the ability to selectively target genes and create deletions, insertions, and base pair substitutions. In this technology, the Cas9 protein is guided towards a sequence with the help of single guide RNA (sgRNA). When using the native Cas9 protein, this results in a specific double‐stranded break (DSB) in the targeted genomic DNA that can be corrected by the native DNA repair mechanisms of the cell[169]. The repair of DSBs is associated with non-specific mutations, since it is typically repaired by the error-prone non-homologous end-joining (NHEJ) pathway, resulting in point mutations, deletions, and frameshifts[170]. To create precise changes in the DNA, the homology-directed repair (HDR) pathway utilizes a DNA repair template which shows homology in the sequences surrounding the DSB. This template can be delivered as a double-stranded DNA template or a single-stranded DNA oligo, which can contain a desired restorative mutation. However, HDR occurs at a relatively low frequency as compared to NHEJ. Thus, in order to generate base pair substitutions without the presence of NHEJ-mediated non-specific mutations, Base Editing (BE) was developed. Here, enzymatic groups that are able to convert specific nucleotides are attached to dCas9, an adapted form of Cas9 whose endonuclease activity is removed through point mutations in its endonuclease domains. As a result, Cas9 will no longer create DSBs while still interacting with specific genomic sequences through the sgRNAs, and the base editing enzymatic groups are able to generate nucleotide substitutions in the sgRNA target sequence. There are different types of base editors, including cytosine base editors (CBEs) and adenine base editors (ABEs) that can mediate single base changes in the genome. Base editors can induce these alterations without requiring DSBs, HDR processes, or donor DNA templates[171-173]. The main drawback of these techniques is that the type of substitutions that can be made are currently limited, and since these substitutions could be made throughout the ~20 nt target sequence of the sgRNA, only a limited number of target sequences are suitable for base editing techniques.
Recently, Anzalone et al. reported a new versatile and precise genome editing method called prime editing[174]. Prime editing does not require a separate donor DNA repair template to precisely edit the DNA. Instead, the needed repair template is incorporated into the sgRNA combined with the desired edit, now called prime editing guide RNA (pegRNA). Furthermore, a catalytically impaired Cas9 nickase is fused to an engineered reverse transcriptase, now called the prime editor (PE). As prime editing is based on the use of a Cas9 nickase, which only cuts one strand of the double helix, DSBs and the subsequent error-prone NHEJ repair process are avoided. After the pegRNA guids the PE towards the target sequence, the reverse transcriptase domain of the PE uses the repair sequence added to the pegRNA to template reverse transcription of the desired edit. This leads to direct transcription of the DNA onto the nicked target DNA strand. The edited DNA strand will replace the original DNA strand, which creates a heteroduplex of one edited strand and one unedited strand. The edited strand will be favored due to the inherent preference of the endogenous endonuclease FEN1 to excise 5’ flaps. This leads to hybridization of the edited 3’ flap being favored copying the edit onto the unedited strand, completing the process. Prime editing can incorporate all the potential 12 modifications; accordingly, prime editing could, in principle, correct up to 89% of known genetic variants associated with human diseases[174,175]. In addition to efficient gene correction in cell lines, prime editing has already been shown to be as efficient in 3D grown organoids derived from patients with inherited metabolic disorders[176]. This substantially expands the scope and capabilities of genome editing, but the in vivo delivery remains challenging[174].
The CRISPR/Cas system can be delivered either as plasmid DNA, mRNA or as ribonucleoprotein (RNP) complex. Delivery of the RNP complex has advantages over plasmid and mRNA delivery, including lower off-target effects, and faster and more efficient gene editing[177]. Nevertheless, there are limitations to in vivo RNP complex delivery, including its large size, negative charge, immunogenicity, pre-existing antibodies[178], and rapid degradation[179]. Loading of the RNP complex into EVs could potentially overcome most of its limitations, as well as lowering its off-target effects by increased tissue targeting.
Whereas EV trafficking towards the lysosome after uptake by recipient cells is an advantage when using engineered EVs to deliver ERT, for the functional delivery of the RNP complex, endosomal escape is required, as the RNP complex needs to be able to reach the cell nucleus to induce genomic editing. Incorporation of viral fusogenic proteins into the EV membrane could increase the relatively low efficiency of endosomal escape of unaltered EVs. The viral protein VSV-G is known to incorporate into the EV membrane upon transfection of the cells with VSV-G encoding plasmids[180]. This protein specifically induces endosomal escape, as it requires the decrease in pH in the late endosome to undergo the conformational transformation that induces the membrane fusion that facilitates cargo release. This will greatly increase endosomal escape and with that the delivery of the EV loaded RNP complexes or other therapeutic components that require delivery to the cytoplasm. Nevertheless, the use of viral proteins can be problematic due to their cytotoxicity and potential immunogenicity[181]. The substitution of these viral proteins is a hurdle that needs to be overcome. EVs may contain some endogenous capacity to escape the endosome that could be explored. For instance, through the incorporation of endogenous proteins with fusogenic activity like syncytins. Syncytins are incorporated into placenta-derived EVs and mediate uptake by recipient cells[182]. Their incorporation into the membrane may facilitate EV fusion and subsequently deliver the EV cargo[129,183]. It is hypothesized that EVs contain additional proteins that facilitate endosomal escape through membrane fusion, as treatment of unmodified EVs with proteinases has been shown to abrogate their capability of membrane fusion[131]. The use of endogenous proteins with fusogenic activity to induce endosomal escape for the delivery of CRISPR/Cas could increase the safety of these EVs.
Efficient gene editing after EV mediated delivery of RNP complexes has already been demonstrated. Active loading can be obtained through direct or indirect fusion of the Cas9 protein to known EV markers. This was shown by the indirect loading approach of Ye et al., who used CD63-GFP and Cas9-GFP nanobody fusion proteins that sufficiently loaded and delivered the complex[184]. Furthermore, Wang et al. proposed the use of ARRDC1, a protein required in plasma membrane budding together with known EV marker TSG101 to form ectosomes[152]. ARRDC1 interacts with proteins containing WW domains and recruits them into vesicles. Wang et al. fused either two or four WW domains to a Cas9 protein and showed that co-expression with ARRDC1 leads to sufficient loading of the RNP complex into EVs[152]. They went on to show functional gene editing upon delivery by knocking down GFP expression in their recipient cells using an anti-GFP sgRNA[152]. Recently, light‐induced dimerization loading was shown to be effective. In this approach, Osteikoetxea et al. used Cryptochrome 2 combined with either CD9 or a Myristoylation‐Palmitoylation‐Palmitoylation lipid modification[185]. This method provides controllable loading and release of the complex upon delivery[185]. In addition to loading of the complex through the Cas9 protein, RNA binding proteins such as HuR can increase complex loading by the addition of AU rich elements to the sgRNA[159]. At the same time, this strategy shows the potential of EV engineering in the delivery of other RNAs, including miRNA[159] and mRNA[152]. Furthermore, the delivery of Cas9-sgRNA or prime editing complexes with EVs could be a major step towards a permanent cure for LSDs. Figure 2 gives an overview of the general EV engineering strategies and the potential of engineered EVs for future LSD treatment.
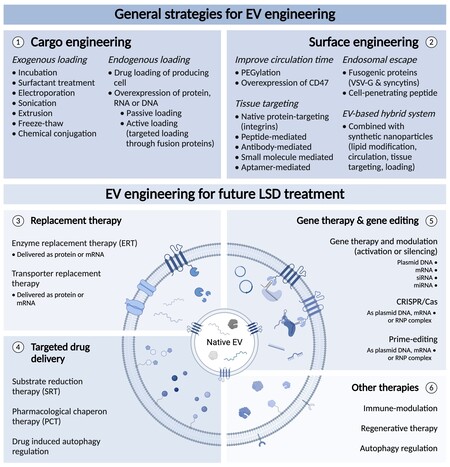
Figure 2. Future EV-based therapeutic options for the treatment of LSDs. Various EV engineering approaches and techniques can be used to optimize the therapeutic potential of EVs, including: (1) Cargo engineering, which can either be accomplished through exogenous loading techniques applied to the EVs post-isolation or endogenous loading techniques. In endogenous loading techniques, loading is accomplished through adaptation of the parental cell. Cargo can either be passively or actively loaded with the use of fusion proteins binding the cargo to EV-enriched proteins or transmembrane domains; (2) Surface engineering, which can improve circulation time of EVs, their tissue targeting potential and endosomal escape, which is needed for efficient intraluminal cargo delivery. Furthermore, EVs can be combined with synthetic nanoparticles. These hybrid systems can modulate the contents and functionality of both the particle surface and cargo. Future EV therapeutic modalities for LSDs potentially include; (3) Replacement therapies, which include enzyme-, and transporter-replacement therapy. These therapies can be loaded into EVs and delivered to target cells as mRNA or directly as protein; (4) Targeted drug delivery, for substrate reduction therapy (SRT), pharmacological chaperon therapy (PCT) and potentially drug induced autophagy regulation; (5) Gene therapy & gene editing, which includes loading of constructs such as plasmid DNA, mRNA, miRNA and siRNA for gene addition or modulation. Moreover, EVs could be engineered to contain CRISPR/Cas or Prime editing constructs in the form of plasmid DNA, mRNA or RNP complexes; and (6) Other therapies, which include unmodified or engineered EVs for adjuvant therapy, including immune-modulatory, regenerative therapy and autophagy regulation.
CONCLUSION
As with any new class of therapeutic agents, EVs face several hurdles in their advancement into the clinic[186]. Optimization of loading efficiency, EV production, isolation, functional properties, purity, storage for potential off-the-shelf availability, as well as selection of the optimal cell source, is needed[47]. In addition, standardization of these processes’ clinical and industry-accepted validation must be developed for the regulatory approval of EV- diagnostics and therapeutics. A better understanding of EV biogenesis, uptake, cargo delivery and the normal in vivo journey will not only accelerate their therapeutic translation, but also improve their function in understanding LSD pathology and use as biomarkers. The EV field holds great potential and may one day contribute to diagnostics and the design of a new generation of smart vehicles for targeted delivery of drugs, ERT, RNA therapeutics and gene editing to manage or treat multisystemic LSDs.
DECLARATIONS
AcknowledgementsFigures were created with BioRender.com.
Authors’ contributionsWrote and edited the manuscript: Hegeman CV, de Jong OG, Lorenowicz MJ
Availability of data and materialsNot applicable.
Financial support and sponsorshipNone.
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Authors 2022.
REFERENCES
1. Walkley SU, Vanier MT. Secondary lipid accumulation in lysosomal disease. Biochim Biophys Acta 2009;1793:726-36.
2. Lamanna WC, Lawrence R, Sarrazin S, Esko JD. Secondary storage of dermatan sulfate in Sanfilippo disease. J Biol Chem 2011;286:6955-62.
3. Prinetti A, Prioni S, Chiricozzi E, Schuchman EH, Chigorno V, Sonnino S. Secondary alterations of sphingolipid metabolism in lysosomal storage diseases. Neurochem Res 2011;36:1654-68.
4. Lieberman AP, Puertollano R, Raben N, Slaugenhaupt S, Walkley SU, Ballabio A. Autophagy in lysosomal storage disorders. Autophagy 2012;8:719-30.
5. Köse S, Aerts-kaya F, Uçkan Çetinkaya D, Korkusuz P. Stem cell applications in lysosomal storage disorders: progress and ongoing challenges. In: Turksen K, editor. Cell biology and translational medicine, Volume 14. Cham: Springer International Publishing; 2021. p. 135-62.
6. Platt FM, Boland B, van der Spoel AC. The cell biology of disease: lysosomal storage disorders: the cellular impact of lysosomal dysfunction. J Cell Biol 2012;199:723-34.
7. Platt FM, d’Azzo A, Davidson BL, Neufeld EF, Tifft CJ. Lysosomal storage diseases. Nat Rev Dis Primers 2018;4:27.
8. Marques ARA, Saftig P. Lysosomal storage disorders - challenges, concepts and avenues for therapy: beyond rare diseases. J Cell Sci 2019;132:jcs221739.
9. Parenti G, Andria G, Ballabio A. Lysosomal storage diseases: from pathophysiology to therapy. Annu Rev Med 2015;66:471-86.
10. Meikle PJ, Hopwood JJ, Clague AE, Carey WF. Prevalence of lysosomal storage disorders. JAMA 1999;281:249-54.
11. Fuller M, Meikle PJ, Hopwood JJ. Epidemiology of lysosomal storage diseases: an overview. In: Mehta A, Beck M, Sunder-Plassmann G, editors. Fabry disease: perspectives from 5 years of FOS. Oxford: Oxford PharmaGenesis; 2006. Chapter 2.
12. Mehta A, Beck M, Sunder-Plassmann G. Fabry disease: perspectives from 5 years of FOS. Oxford: Oxford PharmaGenesis; 2006.
13. Stone WL, Basit H, Master SR. Gaucher disease. StatPearls. Treasure Island (FL): StatPearls Publishing; 2022.
14. Grabowski GA, Dinur T, Osiecki KM, Kruse JR, Legler G, Gatt S. Gaucher disease types 1, 2, and 3: differential mutations of the acid beta-glucosidase active site identified with conduritol B epoxide derivatives and sphingosine. Am J Hum Genet 1985;37:499-510.
15. van den Broek BTA, van Doorn J, Hegeman CV, et al. Hurdles in treating Hurler disease: potential routes to achieve a “real” cure. Blood Adv 2020;4:2837-49.
16. Li M. Enzyme replacement therapy: a review and its role in treating lysosomal storage diseases. Pediatr Ann 2018;47:e191-7.
17. Kishnani PS, Dickson PI, Muldowney L, et al. Immune response to enzyme replacement therapies in lysosomal storage diseases and the role of immune tolerance induction. Mol Genet Metab 2016;117:66-83.
18. Safary A, Akbarzadeh Khiavi M, Mousavi R, Barar J, Rafi MA. Enzyme replacement therapies: What is the best option? Bioimpacts 2018;8:153-7.
19. Solomon M, Muro S. Lysosomal enzyme replacement therapies: historical development, clinical outcomes, and future perspectives. Adv Drug Deliv Rev 2017;118:109-34.
20. Aldenhoven M, Jones SA, Bonney D, et al. Hematopoietic cell transplantation for mucopolysaccharidosis patients is safe and effective: results after implementation of international guidelines. Biol Blood Marrow Transplant 2015;21:1106-9.
21. Guffon N, Pettazzoni M, Pangaud N, et al. Long term disease burden post-transplantation: three decades of observations in 25 Hurler patients successfully treated with hematopoietic stem cell transplantation (HSCT). Orphanet J Rare Dis 2021;16:60.
22. Selvanathan A, Ellaway C, Wilson C, Owens P, Shaw PJ, Bhattacharya K. Effectiveness of early hematopoietic stem cell transplantation in preventing neurocognitive decline in mucopolysaccharidosis type II: a case series. In: Morava E, Baumgartner M, Patterson M, Rahman S, Zschocke J, Peters V, editors. JIMD Reports, Volume 41. Berlin: Springer Berlin Heidelberg; 2018. p. 81-9.
23. Mynarek M, Tolar J, Albert MH, et al. Allogeneic hematopoietic SCT for alpha-mannosidosis: an analysis of 17 patients. Bone Marrow Transplant 2012;47:352-9.
24. Beschle J, Döring M, Kehrer C, et al. Early clinical course after hematopoietic stem cell transplantation in children with juvenile metachromatic leukodystrophy. Mol Cell Pediatr 2020;7:12.
25. Wright MD, Poe MD, DeRenzo A, Haldal S, Escolar ML. Developmental outcomes of cord blood transplantation for Krabbe disease: a 15-year study. Neurology 2017;89:1365-72.
26. Aerts JM, Hollak CE, Boot RG, Groener JE, Maas M. Substrate reduction therapy of glycosphingolipid storage disorders. J Inherit Metab Dis 2006;29:449-56.
27. Hughes DA, Nicholls K, Shankar SP, et al. Oral pharmacological chaperone migalastat compared with enzyme replacement therapy in Fabry disease: 18-month results from the randomised phase III ATTRACT study. J Med Genet 2017;54:288-96.
28. Nagree MS, Scalia S, McKillop WM, Medin JA. An update on gene therapy for lysosomal storage disorders. Expert Opin Biol Ther 2019;19:655-70.
29. Phinney DG, Isakova IA. Mesenchymal stem cells as cellular vectors for pediatric neurological disorders. Brain Res 2014;1573:92-107.
30. Pittenger MF, Discher DE, Péault BM, Phinney DG, Hare JM, Caplan AI. Mesenchymal stem cell perspective: cell biology to clinical progress. NPJ Regen Med 2019;4:22.
31. Jackson M, Derrick Roberts A, Martin E, Rout-Pitt N, Gronthos S, Byers S. Mucopolysaccharidosis enzyme production by bone marrow and dental pulp derived human mesenchymal stem cells. Mol Genet Metab 2015;114:584-93.
32. Hawkins-Salsbury JA, Reddy AS, Sands MS. Combination therapies for lysosomal storage disease: Is the whole greater than the sum of its parts? Hum Mol Genet 2011;20:R54-60.
33. Koç ON, Peters C, Aubourg P, et al. Bone marrow-derived mesenchymal stem cells remain host-derived despite successful hematopoietic engraftment after allogeneic transplantation in patients with lysosomal and peroxisomal storage diseases. Exp Hematol 1999;27:1675-81.
34. Meuleman N, Vanhaelen G, Tondreau T, et al. Reduced intensity conditioning haematopoietic stem cell transplantation with mesenchymal stromal cells infusion for the treatment of metachromatic leukodystrophy: a case report. Haematologica 2008;93:e11-3.
35. de Windt TS, Vonk LA, Slaper-Cortenbach IC, et al. Allogeneic mesenchymal stem cells stimulate cartilage regeneration and are safe for single-stage cartilage repair in humans upon mixture with recycled autologous chondrons. Stem Cells 2017;35:256-64.
36. Yáñez-Mó M, Siljander PR, Andreu Z, et al. Biological properties of extracellular vesicles and their physiological functions. J Extracell Vesicles 2015;4:27066.
37. de Jong OG, Verhaar MC, Chen Y, et al. Cellular stress conditions are reflected in the protein and RNA content of endothelial cell-derived exosomes. J Extracell Vesicles 2012;1:18396.
38. Saint-Pol J, Gosselet F, Duban-Deweer S, Pottiez G, Karamanos Y. Targeting and crossing the blood-brain barrier with extracellular vesicles. Cells 2020;9:851.
39. Banks WA, Sharma P, Bullock KM, Hansen KM, Ludwig N, Whiteside TL. Transport of extracellular vesicles across the blood-brain barrier: brain pharmacokinetics and effects of inflammation. Int J Mol Sci 2020;21:4407.
40. Liu S, Wu X, Chandra S, et al. Extracellular vesicles: emerging tools as therapeutic agent carriers. Acta Pharm Sin B 2022;12:3822-42.
41. Abasolo I, Seras-Franzoso J, Moltó-Abad M, et al. Nanotechnology-based approaches for treating lysosomal storage disorders, a focus on Fabry disease. Wiley Interdiscip Rev Nanomed Nanobiotechnol 2021;13:e1684.
42. Schuh RS, Baldo G, Teixeira HF. Nanotechnology applied to treatment of mucopolysaccharidoses. Expert Opin Drug Deliv 2016;13:1709-18.
43. Grosso A, Parlanti G, Mezzena R, Cecchini M. Current treatment options and novel nanotechnology-driven enzyme replacement strategies for lysosomal storage disorders. Adv Drug Deliv Rev 2022;188:114464.
44. Baixauli F, López-Otín C, Mittelbrunn M. Exosomes and autophagy: coordinated mechanisms for the maintenance of cellular fitness. Front Immunol 2014;5:403.
45. Zaborowski MP, Balaj L, Breakefield XO, Lai CP. Extracellular vesicles: composition, biological relevance, and methods of study. Bioscience 2015;65:783-97.
46. Varderidou-Minasian S, Lorenowicz MJ. Mesenchymal stromal/stem cell-derived extracellular vesicles in tissue repair: challenges and opportunities. Theranostics 2020;10:5979-97.
47. Théry C, Witwer KW, Aikawa E, et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): a position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J Extracell Vesicles 2018;7:1535750.
48. Thakur BK, Zhang H, Becker A, et al. Double-stranded DNA in exosomes: a novel biomarker in cancer detection. Cell Res 2014;24:766-9.
49. Coutinho MF, Prata MJ, Alves S. A shortcut to the lysosome: the mannose-6-phosphate-independent pathway. Mol Genet Metab 2012;107:257-66.
50. Fedele AO, Isenmann S, Kamei M, et al. Lysosomal N-acetyltransferase interacts with ALIX and is detected in extracellular vesicles. Biochim Biophys Acta Mol Cell Res 2018;1865:1451-64.
51. Li Y, He X, Li Q, et al. EV-origin: Enumerating the tissue-cellular origin of circulating extracellular vesicles using exLR profile. Comput Struct Biotechnol J 2020;18:2851-9.
52. Emmanouilidou E, Melachroinou K, Roumeliotis T, et al. Cell-produced alpha-synuclein is secreted in a calcium-dependent manner by exosomes and impacts neuronal survival. J Neurosci 2010;30:6838-51.
53. Trotta T, Panaro MA, Cianciulli A, Mori G, Di Benedetto A, Porro C. Microglia-derived extracellular vesicles in Alzheimer’s Disease: a double-edged sword. Biochem Pharmacol 2018;148:184-92.
54. Ananbeh H, Vodicka P, Kupcova Skalnikova H. Emerging roles of exosomes in huntington’s disease. Int J Mol Sci 2021;22:4085.
55. Tancini B, Buratta S, Sagini K, et al. Insight into the role of extracellular vesicles in lysosomal storage disorders. Genes (Basel) 2019;10:510.
56. Navarro-Romero A, Montpeyó M, Martinez-Vicente M. The emerging role of the lysosome in parkinson’s disease. Cells 2020;9:2399.
57. Almeida MF, Bahr BA, Kinsey ST. Endosomal-lysosomal dysfunction in metabolic diseases and Alzheimer’s disease. Metabolic and bioenergetic drivers of neurodegenerative disease: neurodegenerative disease research and commonalities with metabolic diseases. Elsevier; 2020. p. 303-24.
58. Porro C, Panaro MA, Lofrumento DD, Hasalla E, Trotta T. The multiple roles of exosomes in Parkinson’s disease: an overview. Immunopharmacol Immunotoxicol 2019;41:469-76.
59. Gassart A, Géminard C, Hoekstra D, Vidal M. Exosome secretion: the art of reutilizing nonrecycled proteins? Traffic 2004;5:896-903.
60. Bernardi S, Balbi C. Extracellular vesicles: from biomarkers to therapeutic tools. Biology (Basel) 2020;9:258.
61. Parada N, Romero-Trujillo A, Georges N, Alcayaga-Miranda F. Camouflage strategies for therapeutic exosomes evasion from phagocytosis. J Adv Res 2021;31:61-74.
62. de Jong OG, Kooijmans SAA, Murphy DE, et al. Drug delivery with extracellular vesicles: from imagination to innovation. Acc Chem Res 2019;52:1761-70.
63. Kooijmans SAA, Fliervoet LAL, van der Meel R, et al. PEGylated and targeted extracellular vesicles display enhanced cell specificity and circulation time. J Control Release 2016;224:77-85.
64. Wiklander OP, Nordin JZ, O'Loughlin A, et al. Extracellular vesicle in vivo biodistribution is determined by cell source, route of administration and targeting. J Extracell Vesicles 2015;4:26316.
66. Schröder BA, Wrocklage C, Hasilik A, Saftig P. The proteome of lysosomes. Proteomics 2010;10:4053-76.
67. Chang NC. Autophagy and Stem Cells: self-eating for self-renewal. frontiers in cell and developmental Biology 2020;8.
68. Song HY, Chien CS, Yarmishyn AA, et al. Generation of GLA-knockout human embryonic stem cell lines to model autophagic dysfunction and exosome secretion in fabry disease-associated hypertrophic cardiomyopathy. Cells 2019;8:327.
69. Bartolomeo R, Cinque L, De Leonibus C, et al. mTORC1 hyperactivation arrests bone growth in lysosomal storage disorders by suppressing autophagy. J Clin Invest 2017;127:3717-29.
70. Brown RA, Voit A, Srikanth MP, et al. mTOR hyperactivity mediates lysosomal dysfunction in Gaucher’s disease iPSC-neuronal cells. Dis Model Mech 2019:12.
71. Gao J, Wei B, de Assuncao TM, et al. Hepatic stellate cell autophagy inhibits extracellular vesicle release to attenuate liver fibrosis. J Hepatol 2020;73:1144-54.
72. Skotland T, Sagini K, Sandvig K, Llorente A. An emerging focus on lipids in extracellular vesicles. Adv Drug Deliv Rev 2020;159:308-21.
73. Gurung S, Perocheau D, Touramanidou L, Baruteau J. The exosome journey: from biogenesis to uptake and intracellular signalling. Cell Commun Signal 2021;19:47.
74. Hessvik NP, Øverbye A, Brech A, et al. PIKfyve inhibition increases exosome release and induces secretory autophagy. Cell Mol Life Sci 2016;73:4717-37.
75. Miranda AM, Lasiecka ZM, Xu Y, et al. Neuronal lysosomal dysfunction releases exosomes harboring APP C-terminal fragments and unique lipid signatures. Nat Commun 2018;9:291.
76. Strauss K, Goebel C, Runz H, et al. Exosome secretion ameliorates lysosomal storage of cholesterol in Niemann-Pick type C disease. J Biol Chem 2010;285:26279-88.
77. Ilnytska O, Jeziorek M, Lai K, Altan-Bonnet N, Dobrowolski R, Storch J. Lysobisphosphatidic acid (LBPA) enrichment promotes cholesterol egress via exosomes in Niemann Pick type C1 deficient cells. Biochim Biophys Acta Mol Cell Biol Lipids 2021;1866:158916.
78. van de Vlekkert D, Demmers J, Nguyen XX, et al. Excessive exosome release is the pathogenic pathway linking a lysosomal deficiency to generalized fibrosis. Sci Adv 2019;5:eaav3270.
79. D’Auria L, Reiter C, Ward E, et al. Psychosine enhances the shedding of membrane microvesicles: Implications in demyelination in Krabbe's disease. PLoS One 2017;12:e0178103.
80. Bhat OM, Li G, Yuan X, et al. Arterial medial calcification through enhanced small extracellular vesicle release in smooth muscle-specific asah1 gene knockout mice. Sci Rep 2020;10:1645.
81. Reiter CR, Rebiai R, Kwak A, et al. The pathogenic sphingolipid psychosine is secreted in extracellular vesicles in the brain of a mouse model of krabbe disease. ASN Neuro 2022;14:17590914221087817.
82. Pituch KC, Moyano AL, Lopez-Rosas A, et al. Dysfunction of platelet-derived growth factor receptor α (PDGFRα) represses the production of oligodendrocytes from arylsulfatase a-deficient multipotential neural precursor cells. J Biol Chem 2015;290:7040-53.
83. Chen FW, Li C, Ioannou YA. Cyclodextrin induces calcium-dependent lysosomal exocytosis. PLoS One 2010;5:e15054.
84. Canonico B, Cesarini E, Salucci S, et al. Defective autophagy, mitochondrial clearance and lipophagy in niemann-pick type B lymphocytes. PLoS One 2016;11:e0165780.
85. Alvarez-Erviti L, Seow Y, Schapira AH, et al. Lysosomal dysfunction increases exosome-mediated alpha-synuclein release and transmission. Neurobiol Dis 2011;42:360-7.
86. Vingtdeux V, Hamdane M, Loyens A, et al. Alkalizing drugs induce accumulation of amyloid precursor protein by-products in luminal vesicles of multivesicular bodies. J Biol Chem 2007;282:18197-205.
87. Raben N, Schreiner C, Baum R, et al. Suppression of autophagy permits successful enzyme replacement therapy in a lysosomal storage disorder--murine Pompe disease. Autophagy 2010;6:1078-89.
88. Spampanato C, Feeney E, Li L, et al. Transcription factor EB (TFEB) is a new therapeutic target for Pompe disease. EMBO Mol Med 2013;5:691-706.
89. Gatto F, Rossi B, Tarallo A, et al. AAV-mediated transcription factor EB (TFEB) gene delivery ameliorates muscle pathology and function in the murine model of Pompe Disease. Sci Rep 2017;7:15089.
90. Manjithaya R, Subramani S. Autophagy: a broad role in unconventional protein secretion? Trends Cell Biol 2011;21:67-73.
91. Klein D, Büssow H, Fewou SN, Gieselmann V. Exocytosis of storage material in a lysosomal disorder. Biochem Biophys Res Commun 2005;327:663-7.
92. Yogalingam G, Bonten EJ, van de Vlekkert D, et al. Neuraminidase 1 is a negative regulator of lysosomal exocytosis. Dev Cell 2008;15:74-86.
93. Hastings C, Vieira C, Liu B, et al. Expanded access with intravenous hydroxypropyl-β-cyclodextrin to treat children and young adults with Niemann-Pick disease type C1: a case report analysis. Orphanet J Rare Dis 2019;14:228.
94. Pergande MR, Kang C, George D, et al. Lipidomic analysis identifies age-disease-related changes and potential new biomarkers in brain-derived extracellular vesicles from metachromatic leukodystrophy mice. Lipids Health Dis 2022;21:32.
95. Batzios SP, Zafeiriou DI, Papakonstantinou E. Extracellular matrix components: an intricate network of possible biomarkers for lysosomal storage disorders? FEBS Lett 2013;587:1258-67.
96. Feltri ML, Weinstock NI, Favret J, Dhimal N, Wrabetz L, Shin D. Mechanisms of demyelination and neurodegeneration in globoid cell leukodystrophy. Glia 2021;69:2309-31.
97. Galvan C, Camoletto PG, Cristofani F, Van Veldhoven PP, Ledesma MD. Anomalous surface distribution of glycosyl phosphatidyl inositol-anchored proteins in neurons lacking acid sphingomyelinase. Mol Biol Cell 2008;19:509-22.
98. Koike T, Ishida G, Taniguchi M, et al. Decreased membrane fluidity and unsaturated fatty acids in Niemann-Pick disease type C fibroblasts. BBA-Mol Basis Dis 1998;1406:327-35.
99. Keller S, Ridinger J, Rupp AK, Janssen JW, Altevogt P. Body fluid derived exosomes as a novel template for clinical diagnostics. J Transl Med 2011;9:86.
100. Lässer C, Alikhani VS, Ekström K, et al. Human saliva, plasma and breast milk exosomes contain RNA: uptake by macrophages. J Transl Med 2011;9:9.
101. Liu H, Yuan W, Pang Q, Xue C, Yan X. Single-particle analysis of tear fluid reveals abundant presence of tissue factor-exposing extracellular vesicles with strong coagulation activity. Talanta 2022;239:123089.
102. Nielsen JE, Honoré B, Vestergård K, et al. Shotgun-based proteomics of extracellular vesicles in Alzheimer’s disease reveals biomarkers involved in immunological and coagulation pathways. Sci Rep 2021;11:18518.
103. Upadhya R, Shetty AK. Extracellular vesicles for the diagnosis and treatment of parkinson’s disease. Aging Dis 2021;12:1438-50.
104. Zhou H, Fernhoff P, Vogt RF. Newborn bloodspot screening for lysosomal storage disorders. J Pediatr 2011;159:7-13.e1.
105. Wang RY, Bodamer OA, Watson MS, Wilcox WR. ACMG Work Group on Diagnostic confirmation of lysosomal storage diseases. lysosomal storage diseases: diagnostic confirmation and management of presymptomatic individuals. Genet Med 2011;13:457-84.
106. Puentes-Tellez MA, Lerma-Barbosa PA, Garzón-Jaramillo RG, et al. A perspective on research, diagnosis, and management of lysosomal storage disorders in Colombia. Heliyon 2020;6:e03635.
107. Han JS, Kim SE, Jin JQ, et al. Tear-derived exosome proteins are increased in patients with thyroid eye disease. Int J Mol Sci 2021;22:1115.
108. van den Broek BTA, van Egmond-Ebbeling MB, Achterberg JA, et al. Longitudinal analysis of ocular disease in children with mucopolysaccharidosis i after hematopoietic cell transplantation. Biol Blood Marrow Transplant 2020;26:928-35.
109. Gelb MH. Newborn screening for lysosomal storage diseases: methodologies, screen positive rates, normalization of datasets, second-tier tests, and post-analysis tools. Int J Neonatal Screen 2018;4:23.
110. Metakids. Welke ziekten zitten er in de hielprik? Available from: https://www.metakids.nl/metabole-ziekten-in-de-hielprik/ [Last accessed on 28 Dec 2022].
111. Iyer NS, Gimovsky AC, Ferreira CR, Critchlow E, Al-Kouatly HB. Lysosomal storage disorders as an etiology of nonimmune hydrops fetalis: a systematic review. Clin Genet 2021;100:493-503.
112. Keller S, Rupp C, Stoeck A, et al. CD24 is a marker of exosomes secreted into urine and amniotic fluid. Kidney Int 2007;72:1095-102.
113. Ebert B, Rai AJ. Isolation and characterization of amniotic fluid-derived extracellular vesicles for biomarker discovery. In: Levy B, editor. Prenatal Diagnosis. New York: Springer; 2019. p. 287-94.
114. Levstek T, Mlinšek T, Holcar M, et al. Urinary extracellular vesicles and their mirna cargo in patients with fabry nephropathy. Genes (Basel) 2021;12:1057.
115. Del Pino M, Andrés A, Bernabéu AÁ, et al. Fabry nephropathy: an evidence-based narrative review. Kidney Blood Press Res 2018;43:406-21.
116. Tatiana S, Stanislav N, Darya K, et al. Altered level of plasma exosomes in patients with Gaucher disease. Eur J Med Genet 2020;63:104038.
117. Lo Curto A, Taverna S, Costa MA, et al. Can Be miR-126-3p a biomarker of premature aging? Cells 2021;10:356.
118. Zahran AM, Elsayh KI, El-Deek SE, El-Baz MA. Oxidative stress, trace elements, and circulating microparticles in patients with Gaucher disease before and after enzyme replacement therapy. Clin Appl Thromb Hemost 2015;21:58-65.
119. Moyano AL, Li G, Boullerne AI, et al. Sulfatides in extracellular vesicles isolated from plasma of multiple sclerosis patients. J Neurosci Res 2016;94:1579-87.
120. Krämer-Albers EM, Bretz N, Tenzer S, et al. Oligodendrocytes secrete exosomes containing major myelin and stress-protective proteins: Trophic support for axons? Proteomics Clin Appl 2007;1:1446-61.
121. Best MG, In 't Veld SGJG, Sol N, Wurdinger T. RNA sequencing and swarm intelligence-enhanced classification algorithm development for blood-based disease diagnostics using spliced blood platelet RNA. Nat Protoc 2019;14:1206-34.
122. Best MG, Sol N, In 't Veld SGJG, et al. Swarm intelligence-enhanced detection of non-small-cell lung cancer using tumor-educated platelets. Cancer Cell 2017;32:238-252.e9.
123. Sol N, Leurs CE, Veld SGI', et al. Blood platelet RNA enables the detection of multiple sclerosis. Mult Scler J Exp Transl Clin 2020;6:2055217320946784.
124. Niel G, Carter DRF, Clayton A, Lambert DW, Raposo G, Vader P. Challenges and directions in studying cell-cell communication by extracellular vesicles. Nat Rev Mol Cell Biol 2022;23:369-82.
125. Kumari A, Yadav SK, Yadav SC. Biodegradable polymeric nanoparticles based drug delivery systems. Colloids Surf B Biointerfaces 2010;75:1-18.
126. Sahay G, Querbes W, Alabi C, et al. Efficiency of siRNA delivery by lipid nanoparticles is limited by endocytic recycling. Nat Biotechnol 2013;31:653-8.
127. Flanagan M, Pathak I, Gan Q, et al. Umbilical mesenchymal stem cell-derived extracellular vesicles as enzyme delivery vehicle to treat Morquio A fibroblasts. Stem Cell Res Ther 2021;12:276.
128. Seras-Franzoso J, Díaz-Riascos ZV, Corchero JL, et al. Extracellular vesicles from recombinant cell factories improve the activity and efficacy of enzymes defective in lysosomal storage disorders. J Extracell Vesicles 2021;10:e12058.
129. Kooijmans SAA, de Jong OG, Schiffelers RM. Exploring interactions between extracellular vesicles and cells for innovative drug delivery system design. Adv Drug Deliv Rev 2021;173:252-78.
130. Heusermann W, Hean J, Trojer D, et al. Exosomes surf on filopodia to enter cells at endocytic hot spots, traffic within endosomes, and are targeted to the ER. J Cell Biol 2016;213:173-84.
131. Bonsergent E, Lavieu G. Content release of extracellular vesicles in a cell-free extract. FEBS Lett 2019;593:1983-92.
132. Coulson-Thomas VJ, Caterson B, Kao WW. Transplantation of human umbilical mesenchymal stem cells cures the corneal defects of mucopolysaccharidosis VII mice. Stem Cells 2013;31:2116-26.
133. Lai RC, Arslan F, Lee MM, et al. Exosome secreted by MSC reduces myocardial ischemia/reperfusion injury. Stem Cell Res 2010;4:214-22.
134. Haney MJ, Klyachko NL, Harrison EB, Zhao Y, Kabanov AV, Batrakova EV. TPP1 delivery to lysosomes with extracellular vesicles and their enhanced brain distribution in the animal model of batten disease. Adv Healthc Mater 2019;8:e1801271.
135. Iglesias DM, El-Kares R, Taranta A, et al. Stem cell microvesicles transfer cystinosin to human cystinotic cells and reduce cystine accumulation in vitro. PLoS One 2012;7:e42840.
136. Thoene J, Goss T, Witcher M, et al. In vitro correction of disorders of lysosomal transport by microvesicles derived from baculovirus-infected Spodoptera cells. Mol Genet Metab 2013;109:77-85.
137. Thoene JG, DelMonte MA, Mullet J. Microvesicle delivery of a lysosomal transport protein to ex vivo rabbit cornea. Mol Genet Metab Rep 2020;23:100587.
138. Haney MJ, Zhao Y, Jin YS, Batrakova EV. Extracellular vesicles as drug carriers for enzyme replacement therapy to treat CLN2 batten disease: optimization of drug administration routes. Cells 2020;9:1273.
139. Luan X, Sansanaphongpricha K, Myers I, Chen H, Yuan H, Sun D. Engineering exosomes as refined biological nanoplatforms for drug delivery. Acta Pharmacol Sin 2017;38:754-63.
140. Li K, Yan G, Huang H, et al. Anti-inflammatory and immunomodulatory effects of the extracellular vesicles derived from human umbilical cord mesenchymal stem cells on osteoarthritis via M2 macrophages. J Nanobiotechnology 2022;20:38.
141. Sly WS, Vogler C, Grubb JH, et al. Enzyme therapy in mannose receptor-null mucopolysaccharidosis VII mice defines roles for the mannose 6-phosphate and mannose receptors. Proc Natl Acad Sci USA 2006;103:15172-7.
142. Roefs MT, Heusermann W, Brans MAD, et al. . Evaluation and manipulation of tissue and cellular distribution of cardiac progenitor cell-derived extracellular vesicles
143. Hoshino A, Costa-Silva B, Shen TL, et al. Tumour exosome integrins determine organotropic metastasis. Nature 2015;527:329-35.
144. Park EJ, Prajuabjinda O, Soe ZY, et al. Exosomal regulation of lymphocyte homing to the gut. Blood Adv 2019;3:1-11.
145. Perez-Hernandez D, Gutiérrez-Vázquez C, Jorge I, et al. The intracellular interactome of tetraspanin-enriched microdomains reveals their function as sorting machineries toward exosomes. J Biol Chem 2013;288:11649-61.
146. Elsharkasy OM, Nordin JZ, Hagey DW, et al. Extracellular vesicles as drug delivery systems: Why and how? Adv Drug Deliv Rev 2020;159:332-43.
147. Lennaárd AJ, Mamand DR, Wiklander RJ, El Andaloussi S, Wiklander OPB. Optimised electroporation for loading of extracellular vesicles with doxorubicin. Pharmaceutics 2021;14:38.
148. Dooley K, McConnell RE, Xu K, et al. A versatile platform for generating engineered extracellular vesicles with defined therapeutic properties. Mol Ther 2021;29:1729-43.
149. Gupta D, Wiklander OPB, Görgens A, et al. Amelioration of systemic inflammation via the display of two different decoy protein receptors on extracellular vesicles. Nat Biomed Eng 2021;5:1084-98.
150. Silva AM, Lázaro-Ibáñez E, Gunnarsson A, et al. Quantification of protein cargo loading into engineered extracellular vesicles at single-vesicle and single-molecule resolution. J Extracell Vesicles 2021;10:e12130.
151. Yim N, Ryu SW, Choi K, et al. Exosome engineering for efficient intracellular delivery of soluble proteins using optically reversible protein-protein interaction module. Nat Commun 2016;7:12277.
152. Wang Q, Yu J, Kadungure T, Beyene J, Zhang H, Lu Q. ARMMs as a versatile platform for intracellular delivery of macromolecules. Nat Commun 2018;9:960.
153. Do MA, Levy D, Brown A, Marriott G, Lu B. Targeted delivery of lysosomal enzymes to the endocytic compartment in human cells using engineered extracellular vesicles. Sci Rep 2019;9:17274.
154. Hasilik A. The early and late processing of lysosomal enzymes: proteolysis and compartmentation. Experientia 1992;48:130-51.
155. Youn SW, Li Y, Kim YM, et al. Modification of cardiac progenitor cell-derived exosomes by miR-322 provides protection against myocardial infarction through Nox2-dependent angiogenesis. Antioxidants (Basel) 2019;8:18.
156. Sun D, Zhuang X, Xiang X, et al. A novel nanoparticle drug delivery system: the anti-inflammatory activity of curcumin is enhanced when encapsulated in exosomes. Mol Ther 2010;18:1606-14.
157. Wei H, Chen J, Wang S, et al. A nanodrug consisting of doxorubicin and exosome derived from mesenchymal stem cells for osteosarcoma treatment in vitro. Int J Nanomedicine 2019;14:8603-10.
158. Escudier B, Dorval T, Chaput N, et al. Vaccination of metastatic melanoma patients with autologous dendritic cell (DC) derived-exosomes: results of thefirst phase I clinical trial. J Transl Med 2005;3:10.
159. Li Z, Zhou X, Wei M, et al. In vitro and in vivo RNA inhibition by CD9-HuR functionalized exosomes encapsulated with miRNA or CRISPR/dCas9. Nano Lett 2019;19:19-28.
160. Lloyd-Evans E, Morgan AJ, He X, et al. Niemann-Pick disease type C1 is a sphingosine storage disease that causes deregulation of lysosomal calcium. Nat Med 2008;14:1247-55.
161. Canfrán-Duque A, Pastor O, Quintana-Portillo R, et al. Curcumin promotes exosomes/microvesicles secretion that attenuates lysosomal cholesterol traffic impairment. Mol Nutr Food Res 2014;58:687-97.
162. Lu B, Ku J, Flojo R, Olson C, Bengford D, Marriott G. Exosome- and extracellular vesicle-based approaches for the treatment of lysosomal storage disorders. Adv Drug Deliv Rev 2022;188:114465.
163. Belhadj Z, He B, Deng H, et al. A combined “eat me/don’t eat me” strategy based on extracellular vesicles for anticancer nanomedicine. J Extracell Vesicles 2020;9:1806444.
164. Komuro H, Kawai-Harada Y, Aminova S, et al. Engineering extracellular vesicles to target pancreatic tissue in vivo. Nanotheranostics 2021;5:378-90.
165. Kamerkar S, LeBleu VS, Sugimoto H, et al. Exosomes facilitate therapeutic targeting of oncogenic KRAS in pancreatic cancer. Nature 2017;546:498-503.
166. Kieseier BC, Wisniewski KE, Goebel HH. The monocyte-macrophage system is affected in lysosomal storage diseases: an immunoelectron microscopic study. Acta Neuropathol 1997;94:359-62.
167. Limoni SK, Moghadam MF, Moazzeni SM, Gomari H, Salimi F. Engineered exosomes for targeted transfer of siRNA to HER2 positive breast cancer cells. Appl Biochem Biotechnol 2019;187:352-64.
168. Gomez-Ospina N, Scharenberg SG, Mostrel N, et al. Human genome-edited hematopoietic stem cells phenotypically correct Mucopolysaccharidosis type I. Nat Commun 2019;10:4045.
169. Paquet D, Kwart D, Chen A, et al. Efficient introduction of specific homozygous and heterozygous mutations using CRISPR/Cas9. Nature 2016;533:125-9.
170. Hsu PD, Lander ES, Zhang F. Development and applications of CRISPR-Cas9 for genome engineering. Cell 2014;157:1262-78.
171. Gaudelli NM, Komor AC, Rees HA, et al. Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Nature 2017;551:464-71.
172. Komor AC, Kim YB, Packer MS, Zuris JA, Liu DR. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 2016;533:420-4.
173. Kim YB, Komor AC, Levy JM, Packer MS, Zhao KT, Liu DR. Increasing the genome-targeting scope and precision of base editing with engineered Cas9-cytidine deaminase fusions. Nat Biotechnol 2017;35:371-6.
174. Anzalone AV, Randolph PB, Davis JR, et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 2019;576:149-57.
176. Schene IF, Joore IP, Oka R, et al. Prime editing for functional repair in patient-derived disease models. Nat Commun 2020;11:5352.
177. Liang Y, Iqbal Z, Wang J, et al. Cell-derived extracellular vesicles for CRISPR/Cas9 delivery: engineering strategies for cargo packaging and loading. Biomater Sci 2022;10:4095-106.
178. Simhadri VL, McGill J, McMahon S, Wang J, Jiang H, Sauna ZE. Prevalence of pre-existing antibodies to CRISPR-associated nuclease Cas9 in the USA population. Mol Ther Methods Clin Dev 2018;10:105-12.
179. Gee P, Lung MSY, Okuzaki Y, et al. Extracellular nanovesicles for packaging of CRISPR-Cas9 protein and sgRNA to induce therapeutic exon skipping. Nat Commun 2020;11:1334.
180. Meyer C, Losacco J, Stickney Z, Li L, Marriott G, Lu B. Pseudotyping exosomes for enhanced protein delivery in mammalian cells. Int J Nanomedicine 2017;12:3153-70.
181. Tomás HA, Mestre DA, Rodrigues AF, Guerreiro MR, Carrondo MJT, Coroadinha AS. Improved GaLV-TR glycoproteins to pseudotype lentiviral vectors: impact of viral protease activity in the production of LV pseudotypes. Mol Ther Methods Clin Dev 2019;15:1-8.
182. Vargas A, Zhou S, Éthier-Chiasson M, et al. Syncytin proteins incorporated in placenta exosomes are important for cell uptake and show variation in abundance in serum exosomes from patients with preeclampsia. FASEB J 2014;28:3703-19.
183. Prada I, Meldolesi J. Binding and fusion of extracellular vesicles to the plasma membrane of their cell targets. Int J Mol Sci 2016;17:1296.
184. Ye Y, Zhang X, Xie F, et al. An engineered exosome for delivering sgRNA: Cas9 ribonucleoprotein complex and genome editing in recipient cells. Biomater Sci 2020;8:2966-76.
185. Osteikoetxea X, Silva A, Lázaro-Ibáñez E, et al. Engineered Cas9 extracellular vesicles as a novel gene editing tool. J Extracell Vesicles 2022;11:e12225.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Hegeman CV, de Jong OG, Lorenowicz MJ. A kaleidoscopic view of extracellular vesicles in lysosomal storage disorders. Extracell Vesicles Circ Nucleic Acids 2022;3:393-421. http://dx.doi.org/10.20517/evcna.2022.41
AMA Style
Hegeman CV, de Jong OG, Lorenowicz MJ. A kaleidoscopic view of extracellular vesicles in lysosomal storage disorders. Extracellular Vesicles and Circulating Nucleic Acids. 2022; 3(4): 393-421. http://dx.doi.org/10.20517/evcna.2022.41
Chicago/Turabian Style
Hegeman, Charlotte V., Olivier G. de Jong, Magdalena J. Lorenowicz. 2022. "A kaleidoscopic view of extracellular vesicles in lysosomal storage disorders" Extracellular Vesicles and Circulating Nucleic Acids. 3, no.4: 393-421. http://dx.doi.org/10.20517/evcna.2022.41
ACS Style
Hegeman, CV.; de Jong OG.; Lorenowicz MJ. A kaleidoscopic view of extracellular vesicles in lysosomal storage disorders. Extracell. Vesicles. Circ. Nucleic. Acids. 2022, 3, 393-421. http://dx.doi.org/10.20517/evcna.2022.41
About This Article
Copyright

Data & Comments
Data
0


 Cite This Article 15 clicks
Cite This Article 15 clicks

 Like This Article 31
likes
Like This Article 31
likes







Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.