
DNA damage-induced nuclear factor-kappa B activation and its roles in cancer progression


Abstract
DNA damage is a vital challenge to cell homeostasis. Cellular responses to DNA damage (DDR) play essential roles in maintaining genomic stability and survival, whose failure could lead to detrimental consequences such as cancer development and aging. Nuclear factor-kappa B (NF-κB) is a family of transcription factors that plays critical roles in cellular stress response. Along with p53, NF-κB modulates transactivation of a large number of genes which participate in various cellular processes involved in DDR. Here the authors summarize the recent progress in understanding DNA damage response and NF-kB signaling pathways. This study particularly focuses on DNA damage-induced NF-κB signaling cascade and its physiological and pathological significance in B cell development and cancer therapeutic resistance. The authors also discuss promising strategies for selectively targeting this genotoxic NF-κB signaling aiming to antagonize acquired resistance and resensitize refractory cancer cells to cytotoxic treatments.
Keywords
Introduction
The genome of all living organisms is constantly threatened by a variety of agents which cause DNA damage. DNA lesions may occur by altering DNA bases (i.e. O6-methylguanine and thymine glycols), creating breaks on DNA backbone, and forming cross-links between DNA strands and proteins. Failure to repair these lesions can lead to genomic instability and detrimental consequences.[1] Breaks on both strands of DNA (double-stranded break, DSB) represent one of the most lethal types of genomic lesion, which has been associated with pathogenesis of a variety of human diseases and aging.[2] DSB can be induced by environmental exposure such as ultraviolet (UV) or ionizing radiation (IR), as well as by endogenous agents like reactive oxygen species generated by cell metabolism.[3] Genomic lesions can be recognized and labeled by recruitment of sensor proteins, which activates a complex network of cellular responses known as DNA damage response (DDR) and mobilizes DNA repair machinery in order to maintain genomic integrity.[4] Low levels of DNA damage cause cell cycle arrest and promote repair of DNA lesions, whereas severe DNA damage leads to apoptosis or permanent cell cycle arrest (senescence) to avoid neoplastic transformation.[5] DDR is often deregulated in malignant cells, which allows them to escape apoptosis or senescence. These cells could proliferate while harboring DNA lesions, which significantly increases the chance of genetic mutation. A number of anti-apoptotic signaling pathways, such as nuclear factor kappa B (NF-κB), have been shown to also play critical roles in modulating cancer cell response to DNA damage.[6]
NF-κB is a family of transcription factors that play critical roles in inflammation, immunity, cell proliferation, development, survival and apoptosis.[7-9] The inactive NF-κB is present in the cytoplasm in most cell types and it can be activated by a variety of extra-cellular stimuli such as pro-inflammatory cytokines, bacterial lipopolysaccharides, and viral RNA and DNA, via the activation of membrane and cytosolic receptors.[10,11] NF-κB was also shown to be activated by DNA damaging drugs in a membrane receptor-independent manner, which involves a retrograde signaling cascade from nucleus to cytoplasm.[6,12-14] It has been reported that NF-κB was activated in response to a variety of DNA lesions, such as temozolomide-induced SN1-methylation,[15] cisplatin-induced DNA cross-linking,[16] and IR-induced DSB. Recent studies have revealed a variety of roles of DNA damage-activated NF-κB in cancer cell responses to radiation and chemotherapies as well as in cancer progression and metastasis. This review will focus on the recent progress in understanding DNA damage-induced signaling, DDR, and genotoxic DSB agent-induced NF-κB signaling cascade, as well as their physiological functions and pathological significance in cancer progression, therapeutic resistance and metastasis.
DSB and DNA damage signaling
DSB is the most severe form of genomic lesion due to the potential errors incurred during DSB repair. Cells are equipped with several repair mechanisms including homologous recombination (HR), classical non-homologous end joining (cNHEJ), back-up alternative NHEJ (aNHEJ) and single-strand annealing.[4,17-19] Successful HR is generally error-free while NHEJ and other alternative forms of DNA repair are more likely to introduce DNA lesions. Most DSBs are repaired quickly, but those DSBs which repaired incorrectly or escaped repair mechanisms could cause chromosomal aberrations, loss of heterozygosity, oncogenic mutation, or cell death.
Endogenous and exogenous causes of DSBs
DSB can be induced by endogenous molecules such as reactive oxygen species, lipid peroxides, endogenous reactive chemicals (e.g. aldehydes and S-adenosylmethionine), telomere attrition and depurination mechanism.[3] Physiological DSB can also be generated during V(D)J recombination of immunoglobin chains in lymphocytes.[20] Moreover, DSBs are also formed indirectly from collapse of stalled transcription forks or arrested replication forks. These replication fork arrests could occur during normal replication at sequences which are prone to form secondary structures such as tRNA genes and chromosomal fragile sites.[21,22]
Genotoxic agents are present in the environment at a very low level, whereas higher levels can be found in diagnostic tools and tumor therapies. The exogenous causes of DSBs are mostly either accidental exposure or medical procedures. A harmful dose of IR is normally not present in the environment, but such a dose could be received from accidental exposure to radioactive materials or, theoretically, a nuclear attack. IR at a dose of 1 Gy leads to approximately 1,000 SSBs and 20-40 DSBs per cell, among which DSBs are more cytotoxic although less in the number of breaks.[4] Keep in mind that diagnostic imaging techniques such as X-rays and mammograms use a very low level of radiation which could induce DSB directly and indirectly via oxidative stress. In addition, radiation therapy and cytotoxic chemotherapeutic drugs, such as camptothecin, doxorubicin and daunorubicin, induce DSBs in cancer cells through directly damaging DNA or interfering DNA topoisomerase function, leading to apoptosis and elimination of malignant cells.
DNA damage response
Damage to DNA can elicit a complex cellular response by activating multiple signaling cascades, which are generally termed DNA damage response.[4] DNA double-strand breaks could be recognized and bound by a protein complex called MRE11/RAD50/NBS1 (MRN) within a few seconds of their formation. As a DSB lesion sensor, MRN complex binds to the break extremities, stabilizes them close to each other and initiates DDR via NBS1-dependent recruitment of ATM kinase.[23-25] In resting cells, inactive ATM dimer associates with the Tip60 histone acetyltransferase and protein phosphatase 2A.[26,27] Upon DNA damage, MRN complexes bound to DNA breaks recruit Tip60 on histone 3 trimethylated on K9 (H3K9me3). This interaction activates Tip60 which in turn acetylates ATM kinase on K3016.[28,29] ATM acetylation induces its confirmation change and auto phosphorylation on S367, S1893, S1981 and S2996 as well as dimer-to-monomer transition.[30-32] The dissociation of protein phosphatase 2A, which targets phosphorylated S1981, from ATM is also required for full activation of ATM.[27]
Around 10% of activated ATM by irradiation or neocarzinostatin treatment associates with chromatin in the form of ionizing radiation induced foci (IRIF), whereas the majority of active ATM remains free in nucleoplasms.[33,34] As a master regulator of the DSB-induced DDR, ATM phosphorylates various substrates at the consensus target sequence, (S/T)Q, so as to orchestrate the activation of multiple signaling pathways regulating cell cycle arrest, DNA repair, and apoptosis as well as other pathophysiological processes.[35] ATM substrates can be divided into subsets based on their subcellular localization, such as chromatin-associated (H2AX, KAP1), integrated in the IRIF (MDC1, 53BP1, BRCA1, NBS, MRE11, RNF20-RNF40), IRIF-adjacent and phosphorylated by IRIF-bound ATM (Chk2), or phosphorylated by a free-floating pool of ATM (p53, NEMO) in nucleoplasm. Beyond those nuclear substrates, a subset of ATM substrates localized in the cytoplasm (4EBP1, TAB2) has also been reported to play critical roles in cellular response to DSBs.[2,4]
ATM belongs to a family of PI3K-related protein kinases which includes ATM, ATR, DNA-PKcs, mTOR, SMG-1 and TRRAP.[4] Although they all share the similar kinase domain as that in lipid kinase PI3K, they are protein kinases except for TRRAP whose kinase activity remains to be validated. Along with ATM, DNA-PK and ATR also play essential roles in mediating DNA damage response. In human cells, most breaks are rapidly repaired by cNHEJ throughout the entire cell cycle.[17,36] DNA-PKcs is indispensable for repairing DNA double-strand breaks by NHEJ. DNA-PKcs can be recruited to DSBs by the Ku70/Ku80 heterodimer and form the active DNA-PK complex, which promotes synapsis of the broken DNA ends.[37] Like ATM, DNA-PKcs is constitutively associated with Tip60, which controls its activity. Knockdown of Tip60 by siRNA reduces the phosphorylation and activation of DNA-PKcs in response to bleomycin.[38] Most DNA-PK substrates are implicated in DNA repair (DNA-PK itself, Artemis, polynucleotide kinase, XLF, excision repair cross complementing 4), whereas DNA-PK-dependent phosphorylation of H2AX, KAP-1, p53 leads to activation of cell death machinery.[39,40] As a replication stress sensor binding to single-strand DNA, heterotrimeric Replication Protein A (RPA) accumulates at stalled replication forks and recruits ATR interacting protein (ATRIP) in association with ATR kinase. Activation of ATR also requires Rad9/Rad1/Hus1 heterotrimer (9-1-1 complex) and the DNA topoisomerase binding protein 1 (TopBP1).[41] ATR kinase activity is necessary for stabilization and restart of stalled replication forks, and for signaling to cell cycle checkpoint activation.[42,43] Therefore, ATR is essential for cell replication and viability as well as maintaining genomic stability.[44,45]
NF-κB activation in DNA damage response
Besides the prompt cellular responses (e.g. cell cycle arrest, DNA repair) to counteract DNA lesions, transcription/expression of a large number of genes can also be altered in response to DNA damage. Two transcription regulators, p53 and NF-κB, have been identified as the major players for reprogramming the transcription of these genes in response to IR.[46-48] DNA damage-dependent regulation of p53 signaling has been well studied and comprehensively reviewed.[2,49,50] Here we focus on the recent progress in understanding genotoxic stress-induced NF-κB signaling.
NF-κB family
NF-κB is a family of transcription factors composed of five members, p65 (RelA), c-Rel, RelB, p105/p50 and p100/p52, which form hetero- or homo-dimers and regulate a variety of physiological and pathological processes. In resting cells, NF-κB localizes in the cytoplasm in association with a family of inhibitor proteins called IκBs (inhibitor of NF-κB), such as IκBα.[11,51] Upon stimulation, NF-κB is released from IκBs and translocates into the nucleus, where it binds to the promoter and/or enhancer regions of its target genes and regulates their transcription. In addition to nuclear translocation, posttranslational modification of NF-κB, such as phosphorylation, acetylation and methylation of p65, also plays a significant role in modulating transcriptional activity.[11,52] A large number of NF-κB-target genes have been identified (see list at www.nf-kb.org), which participate in a wide range of physiological and pathological processes, such as cell proliferation, innate and adaptive immune responses, inflammation, cell migration, and regulation of apoptosis, among others.[11,53]
Classical and alternative NF-κB signaling pathways
Previous studies have established two well-defined NF-κB activation signaling pathways initiated from membrane-bound receptors, the so-called “classical” and “alternative” pathways.[54] The classical NF-κB pathway depends on activity of the IKK (IκB kinase) kinase complex, which is composed of IKKα, IKKβ and IKKγ/NEMO. Upon activation of the IKK complex, the IKKβ subunit directly phosphorylates NF-κB-associated IκBα, leading to its proteasomal degradation and release of p65/p50 heterodimer. Free NF-κB then translocates into the nucleus and regulate gene transcription. The alternative pathway of NF-κB activation relies on the IKKα homodimer activation in a manner dependent on NF-κB inducing kinase (NIK). Activated IKKα then phosphorylates p100 and promotes partial processing of p100 and yielding of p52. Consequent p52: RelB dimmer then translocates into nucleus and regulate the transcription of its target genes.
DNA damage-induced NF-κB signaling pathway
DNA-damaging agents also activate NF-κB in a canonical IKK complex-dependent fashion. However, in contrast to classical or alternative NF-κB signaling pathways, this genotoxic signaling cascade is initiated in the nucleus instead of via membrane-bound receptors. In the following section, we will discuss the detailed molecular signaling events mediating this retrograde signaling pathway [Figure 1].[6,14]
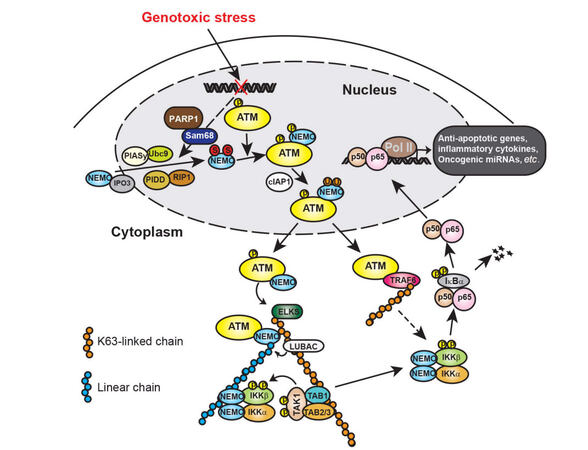
Figure 1. Illustration of genotoxic stress-induced NF-κB signaling cascades. In response to genotoxic treatments, NEMO translocated into nucleus could be SUMOylated by PIASy, which enhances the nuclear accumulation of NEMO. The SUMOylation of NEMO may be facilitated by PARP-1/Sam68 and/or PIDD/RIP complex. Nuclear accumulated NEMO can further form a complex with ATM that phosphorylates NEMO and promotes NEMO monoubiqutination. Monoubiquitinated NEMO then exports into cytoplasm along with ATM, where they form a complex with ELKS. ATM-promoted ELKS ubiquitination with K63 chains recruits LUBAC complex, which facilitates the assembly of linear ubiquitin chain attached on NEMO. The ELKS/NEMO anchored ubiquitin chains stabilize binding of TAK1 and IKK complex thereby promoting their activation. ATM may also export into cytoplasm and form a complex with TRAF6, which leads to TRAF6 polyubiquitination. The polyubiquitin chains attached on TRAF6 could also enhance IKK activation. Activated IKK then phosphorylates IκBα and frees NF-κB for nuclear translocation. In the nucleus, NF-κB could drive transactivation of anti-apoptotic genes (e.g. Bcl-xL, XIAP and Survivin), inflammatory cytokines (e.g. IL-6 and IL-8) and oncogenic miRNAs (e.g. miR-21 and miR-181a), resulting in therapeutic resistance and aggressive metastasis in cancer cells. NF-κB: nuclear factor kappa B; PIASy: protein inhibitor of activated; PARP-1: poly (ADP-ribose) polymerase 1; PIDD: p53-induced death domain protein; RIP: receptor interacting protein; TAK1: TGF-beta activated kinase; IKK: IκB kinase 1; TRAF6: TNF receptor-associated factor 6
Nuclear steps: ATM phosphorylates SUMOylated NEMO
ATM is the pivotal kinase involved in NF-κB activation following DNA damage. In 1998, Lee et al.[55] first observed that NF-κB activation by IR was reduced in human cells with ATM deficiency (A-T cells). Later Piret et al.[56] reported that decreased NF-κB activation by the chemotherapeutic drug CPT in A-T cells could be restored by complementation with ATM. Moreover, both IR and NCS treatment induced ATM-dependent IKKβ activation in HEK293 cells.[46] Therefore, the activation of ATM by DNA damage likely serves as a cornerstone to bridge nuclear DNA damage response to cytoplasmic activation of NF-κB signaling cascade.
The IKK kinase complex is the core component of the classical NF-κB cascade which is also required for DNA damage induced NF-κB activation. The non-catalytic subunit of the IKK complex, NEMO, was found to play unique roles in mediating genotoxic NF-κB activation which may be dispensable in classical NF-κB signaling. For example, the C-terminal zinc finger (ZF) domain of NEMO was shown to be essential for NF-κB activation following treatment with DNA-damaging agents. In contrast, NEMO ZF domain deletion minimally affected NF-κB activation following treatment with canonical stimuli (e.g. lipopolysaccharide). Importantly, a small fraction of NEMO was found to disassociate from IKKα/β upon DNA damage and translocate into nucleus by association with IPO3 (importin 3, transportin 2).[57] Subsequently, nuclear NEMO can be modified by a small protein called SUMO (small ubiquitin-like modifier) in the nucleus in response to DNA damage.[58] Similar to ubiquitin, SUMO can be covalently conjugated onto lysine residue of its target proteins and thereby altering the function and activity of the SUMOylated proteins. SUMO E3 ligase PIASy (protein inhibitor of activated STATy)-mediated SUMOylation on K277 and 309 of NEMO enhanced its nuclear accumulation which is essential for subsequent signaling events activating NF-κB upon DNA damage.[58,59] Nevertheless, the mechanism through which SUMOylated NEMO is localized to nucleus is still unclear.
Two different protein complexes have been shown to regulate the NEMO SUMOylation. Subsequent to DNA damage, p53-induced death domain protein (PIDD) and receptor interacting protein 1 (RIP1) associate with nuclear NEMO as a heterotrimer and accumulate in the nucleus. This PIDD/RIP1/NEMO complex may promote NEMO SUMOylation following chemotherapeutic drug treatment in HEK293 cells.[60] The second modulator of NEMO SUMOylation is poly (ADP-ribose) polymerase 1 (PARP-1). PARP-1 is an abundant chromatin-associated enzyme that can be quickly recruited to sites of SSB and DSB as a DNA damage sensor. After recruitment to the breaks, PARP-1 is activated by post-translational modifications by adding poly (ADP-ribose) to acceptor proteins such as itself and histones. PARylation alters the steric properties of the PARylated proteins, leading to change of their interacting partners. PARP-1 is essential for maintaining genomic integrity and involved in base excision repair, SSB and DSB repair, DNA methylation, transcription regulation, and also signal transduction.[61-63] DNA-bound PARylated PARP-1, or free PARylated PARP-1 in the nucleoplasm, serves as a docking platform for several proteins, such as PIASy.[64,65] Upon DNA damage, PARylated PARP-1 was found to form a transient nuclear signalsome along with ATM, NEMO and PIASy, and PIASy binding to active PARylated PARP1 is essential for DNA damage-induced NEMO SUMOylation and nuclear accumulation.[65] A recent study also identified Src-associated-substrate-during-mitosis-of-68 kDa/KH domain containing RNA binding, signal transduction-associated 1 (Sam68/KHDRBS1) as a key NF-κB regulator in the genotoxic stress-initiated NF-κB signaling pathway.[66] Sam68 deficiency abolished DNA damage-induced PARylation and the PARP1-dependent NF-κB-mediated transactivation of anti-apoptotic genes. Consistently, Sam68 deficient cells are hypersensitive to genotoxic treatment while overexpression of Sam68 elevated PAR production and NF-κB-mediated anti-apoptotic transcription in colon cancer cells. Another study suggested that cell membrane protein MUC13 may also participate in regulation of genotoxic NF-κB signaling. Although the detailed mechanism is still unclear, it may involve stabilization of PARP1, enhanced ATM phosphorylation and NEMO SUMOylation.[67]
In cells exposed to genotoxic treatments, increased nuclear localization of NEMO substantially enhances its association with ATM.[68] Furthermore, PARP-1 may also stabilize the interaction between NEMO and ATM through PARylation of ATM and formation of the aforementioned nuclear signalsome.[65] The association between NEMO and activated ATM leads to ATM-dependent phosphorylation of NEMO on Ser 85.[68] The precise function of NEMO phosphorylation remains to be determined, but ATM activity and intact NEMO-Ser85 are prerequistites for subsequent NEMO monoubiquitination, suggesting NEMO phosphorylation on Ser85 may serve as a cue for its subsequent ubiquitination, such as cIAP1 recruitment. E3 ubiquitin ligase cIAP1, was shown to mediate NEMO mono-ubiquitination at K277 and 309 in the nucleus upon DNA damage in an ATM-dependent manner.[69] cIAP1 may compete with SUMO ligase PIASy for NEMO association, as both bind to the same region of NEMO and target the same residues. When overexpressed, cIAP1 inhibits NEMO SUMOylation. Although the function of NEMO monoubiquitination is not fully elucidated, various studies suggested that ubiquitination also regulates subcellular localization of NEMO. In contrast to SUMOylation, monoubiquitination appears to promote nuclear export of NEMO so as to transduce a nuclear signaling into cytoplasm and relay to downstream signaling events.
The nuclear export of NEMO is essential to convey the signal from nucleus to cytoplasm and the underlying mechanisms have been only partially elucidated. Huang et al.[58,68] demonstrated that NEMO monoubiquitination is required for its Ca2+- and Ran-GTP-dependent nuclear export. ATM and NEMO are found to be exported together. Another study showed that NEMO may be monoubiquitinated in the cytoplasm after the export of SUMOylated NEMO from the nucleus.[70] ATM may be also exported in a Ca2+-dependent but PARP-1/NEMO/PIASy-independent manner. Nevertheless, the presence of monoubiquitinated NEMO in cytoplasm delivers the nuclear DNA damage signal to cytoplasmic compartment and leads to a cytoplasmic NEMO: ATM: IKK complex, which further promotes NF-κB activation.[21]
Cytoplasmic steps: ATM mediates TAK1-IKK activation
In the cytoplasm, ATM still plays important roles to activate the IKK complex. NEMO and ATM were found to form a complex with the IKK-associated protein ELKS (a protein rich in glutamate, leucine, lysine and serine, also called ERC1).[68] ELKS has been shown to play a role in synaptic plasticity, intracellular transport, and exocytosis by regulating release of neurotransmitters at presynaptic active zones.[71-73] ELKS has been found as a putative IKK complex component regulating IKK-dependent IκBα phosphorylation in TNFα-induced NF-κB activation.[74] ELKS also forms a complex with NEMO, ATM and IKK, which is required for activation of IKK-upstream kinase TGF-beta activated kinase 1 (TAK1).[68,75] Further investigation revealed that ELKS is conjugated with K63-linked polyubiquitin chains which depends on ATM and ubiquitin ligase XIAP. Moreover, ATM may also directly bind and promote the ubiquitin ligase activity of TNF receptor-associated factor 6 (TRAF6), leading to TRAF6 auto-ubiquitination with K63-chains.[70] The K63-linked polyubiquitin chains conjugated on ELKS and TRAF6 could then serve as a docking platform of TAK1/TAB1/TAB2 complex and lead to its activation.
Besides K63-linked polyubiquitination, linear ubiquitin chains also contribute to NF-κB activation by DNA damage.[76] Linear ubiquitin chains are connected by a head-to-tail peptide bond between C-terminal Gly76 of one ubiquitin and N-terminal α-amino group on Met1 of another ubiquitin molecule.[77] The LUBAC protein complex comprised of hemeoxidized IRP2 ubiquitin ligase-1 (HOIL1), HOIL1-interacting protein (HOIP) and shank-associated RH domain interactor (SHARPIN), was identified as the only E3 ligase specifically to promote linear ubiquitin chain formation.[78-81] Upon TNFα stimulation, LUBAC was found in a TNFR-supercomplex where it facilitates linear ubiquitination of RIP1 and NEMO.[82,83] The UBAN domain of NEMO has high affinity for interaction with the linear ubiquitin chain,[84,85] suggesting the linear ubiquitin chains attached on NEMO or RIP1 may be stabilizing the NEMO/IKK complex within the TNFR-supercomplex, leading to effective activation of IKK. Similarly, NEMO could be modified by linear ubiquitin chains in the cytoplasm of cells exposed to DNA damage. LUBAC is required for DNA damage-induced NEMO linear ubiquitination.[76]
The linear chain-conjugated lysine residues in NEMO have been identified as Lys285 and Lys309. Lys309 could be modified by monoubiquitination and Lys285 was shown to be conjugated with a single ubiquitin moiety upon DNA damage in another study.[70] Therefore, it is likely the mono-ubiquitin attached on Lys285 and 309 may serve as a cornerstone for further extension of linear ubiquitin chains. The K63 chains attached on ELKS and TRAF6, along with linear chains anchored on NEMO, may form an intertwined network which provides an optimal binding platform for recruiting and stabilizing association of TAK1/TAB1/TAB2 and IKK complexes. The clustering of TAK1 and IKK complexes leads to effective auto-phosphorylation and activation of TAK1 and subsequent TAK1-dependent IKK activation upon DNA damage. After IKK activation, the downstream signaling events are similar to that in the classical NF-κB signaling cascade, which involves IKK-dependent IκBα phosphorylation and degradation, free NF-κB nuclear translocation and target gene transcription alteration.
In addition to the well-described mechanistic connection linking DNA damage signals to the canonical IKK-NF-κB pathway, DNA damage may also lead to activation of alternative NF-κB pathways. RelB was found to be enriched in the nuclei following ionizing radiation in prostate cancer cells, which correlated with poor prognosis in prostate cancer patients.[86-88] In osteosarcoma cell lines, p100 phosphorylation and subsequent processing to p52 observed following multiple forms of DNA damage. However, how DNA damage activates alternative NF-κB pathway, and the mechanistic roles of IKKα, NEMO or ATM in this genotoxic signaling cascade, still remain to be elucidated.
Resolution of DNA damage-induced NF-κB activation
As NF-κB activation plays critical roles in both physiological (e.g. immunity, cell proliferation and survival) and pathological (e.g. inflammation, auto-immune response and cancer progression) processes, tight control of NF-κB activation is essential for maintaining homeostasis of cell functions. Negative feedback regulation assumes an important part in the control of NF-κB activity.[89,90] A classic example is NF-κB-dependent induction of IκBα synthesis after stimulation, which specifically antagonizes NF-κB activity and prevents prolonged NF-κB activation.[91,92] Cells deficient in IκBα present basal, and more sustained signal-inducible, NF-κB activities.[93]
Another shared negative feedback mechanism relies on an induced inhibitory mechanism targeting NF-κB-activating signaling events. Recent studies have shown that expression of deubiquitinases (DUBs), such as A20, are prompted by TNFα stimulation in an NF-κB-dependent manner. These DUBs then cleave polyubiquitin chains to limit IKK activation.[94-97] Thus, a deficiency in DUBs A20 or CYLD (cylindromatosis) can prompt augmented and sustained NF-κB activity in response to inflammatory stimuli and lead to inflammatory disorders as well as oncogenesis. In the following section, we focus on the negative feedback mechanisms induced by DNA damage, which limit the genotoxic NF-κB activation [Figure 2].
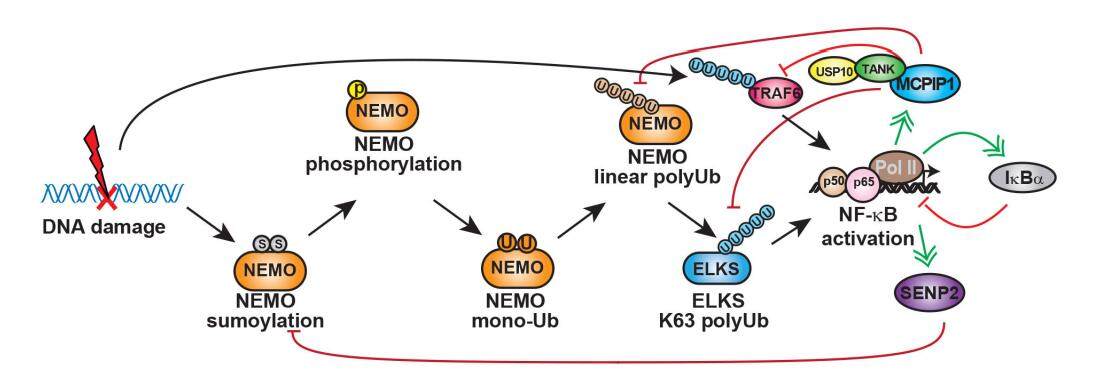
Figure 2. Negative feedback mechanisms modulating genotoxic NF-κB activation. Upon genotoxic NF-κB activation, desumoylation enzyme SENP2 can be transcriptionally upregulated, which in turn decreases NEMO sumoylation and suppresses genotoxic NF-κB signaling. Similarly, MCPIP1 can be upregulated by NF-κB in response to genotoxic treatment. MCPIP1 may decrease NEMO linear ubiquitination and ELKS K63 polyubiquitination by facilitating their interaction with USP10. Meanwhile, MCPIP1/USP10 forms a complex with TANK, which bridges the association of the deubiquitinase complex with TRAF6 and suppresses TRAF6 ubiquitination. All these deubiquitination events could contribute to the abrogation of genotoxic NF-κB activation. In addition, as a canonical NF-κB target gene, IκBα induction could also diminish NF-κB activation by DNA damage. NF-κB: nuclear factor kappa B; SENP2: Sentrin/SUMO-specific protease 2; MCPIP1: monocyte chemotactic protein-1-induced protein-1; TANK: TRAF family member-associated NF-κB activator; TRAF6: TNF receptor-associated factor 6
SENP2-dependent inhibition of NF-κB activation by DNA damage
In DNA damage signaling, SUMOylation of NEMO is a critical signaling event in mediating IKK and NF-κB activation.[58] SUMOylation is the posttranslational modification of lysine residues in target proteins by covalent connection of a SUMO moiety, and is biochemically analogous to, but functionally distinct from, ubiquitination. Like DUBs restricting ubiquitination, individuals from the Sentrin/SUMO-specific protease (SENP) family remove SUMO conjugates from their substrates.[98,99] Due to the reversible nature of SUMOylation, it has been tempting to speculate that desumoylation by an inducible SENP may negatively regulate genotoxic NF-κB activation. Indeed, it was found the SENP2 and SENP1 are major and minor negative regulators, respectively, of NF-κB signaling induced by genotoxic stimuli.[100] Among the six human SENPs, SENP2 interacted most efficiently with NEMO and robustly attenuated NF-κB activation by genotoxic stress. SENP2 overexpression decreased the level of NEMO SUMOylation and NF-κB activation initiated by DNA damage. While wild-type MEFs demonstrated transient NF-κB activation, Senp2−/−MEFs indicated increased genotoxic stress-instigated NEMO SUMOylation and NF-κB activation.[100] More interestingly, SENP2 and SENP1 genes are direct targets of NF-κB whose transcription was substantial increased upon DNA damage. Chromatin IP analysis indicated that treatment with the genotoxic drug etoposide, but not TNFα, leads to increased H3K4me2 at the SENP2 promoter, an epigenetic marker associated with active transcription.[101,102] The induced histone methylation was ATM-dependent, which is consistent with previous findings that ATM regulates telomere elongation through the H3K4 methyltransferase SpSet1p.[103] Moreover, ATM was shown to promote DNA repair by regulating RNF20 phosphorylation and histone methylation, including H3K4me2.[104,105] These results also indicate that ATM not only mediates activation of NF-κB signaling in response to DNA damage, it also facilitates NF-κB-dependent transcription by promoting epigenetic modification to synergistically enhance NF-κB-target gene induction. As in the situation with DUBs in the cytokine signaling,[106] the relative significance of NF-κB-dependent feedback regulation mediated by SENPs in genotoxic signaling probably depends on cell types and the nature of DNA damage stimuli. Nevertheless, SENP2/1 induction upon genotoxic stress provided the first negative-feedback mechanism to uniquely regulate DNA damage-induced NF-κB activation through modulating SUMOylation/desumoylation of NEMO.
Suppression of genotoxic NF-κB activation by MCPIP1/USP10
Monocyte chemotactic protein-1-induced protein-1 (MCPIP1, also called ZC3H12A) was initially recognized as a potential transcription factor in cardiac myocytes regulating apoptosis and chronic inflammatory response.[107] Further studies demonstrated that MCPIP1 can be induced in macrophages, which depend on down-regulation of NF-κB signaling, to modulate inflammatory gene expression. MCPIP1-knockout mice displayed severe immune disorders, growth retardation and premature death.[108-110] It was found that MCPIP1 could bind to the 3’-untranslated regions (UTRs) of a subset of inflammatory cytokine genes including IL6 and IL12, and destabilize the bound mRNAs through its RNase activity.[110] Intriguingly, MCPIP1 was found to limit LPS-induced NF-κB activation in macrophages through expulsion of polyubiquitin chains from TRAF proteins.[109,111] The transcription of MCPIP1 was also upregulated, in a NF-κB-dependent fashion, in cells exposed to genotoxic stimulation.[112] Further investigation revealed that MCPIP1, although itself lacks DUB activity, could serve as an adaptor protein to enhance interaction of deubiquitinase USP10 with polyubiquitinated NEMO, which in turn removes the linear ubiquitin chains from NEMO and suppresses NF-κB signaling upon DNA damage. Two N-terminal domains of MCPIP1 are essential for MCPIP1 to direct USP10-dependent deubiquitination. The RNase-CCCH domain is required for interaction between MCPIP1/NEMO/USP10 and the UBA domain is required for MCPIP1 binding to ubiquitin chains, which may present the ubiquitinated substrates to USP10 for cleavage.[112] Consistently, NF-κB-dependent gene transcription upon DNA damage was significantly enhanced in MCPIP1-deficient cells. Therefore, induction of MCPIP1 serves as a negative feedback response to attenuate NF-κB activation by DNA damage.
TANK-dependent inhibition of NF-κB signaling
TRAF family member-associated NF-κB activator (TANK, also known as I-TRAF) was originally identified as a protein associated with the TRAF2 and TRAF3, and involved in TRAF-mediated NF-κB signaling pathways.[113-115] In response to viral infection-induced retinoic acid-inducible gene 1 (RIG-I) activation, TANK serves as an adaptor bridging TRAF3 association with TBK1 and IKKϵ, which promotes phosphorylation and activation of Interferon Regulatory Factor 3 (IRF3)/IRF7 as well as NF-κB signaling.[116-119] However, TANK was also shown to negatively regulate NF-κB activation.[119,120] NF-κB activation upon TLR or BCR (B-cell receptor) stimulation was enhanced in macrophages and B cells isolated from Tank−/− mice compared with their wild-type counterparts. Interestingly, TANK deficiency increased TRAF6 ubiquitination in response to TLR stimulation in macrophages, which may account for the increased NF-κB activation. However, no canonical deubiquitination enzyme domain can be found in TANK. Also, neither A20 nor CYLD, two common DUBs involved in negative regulation of NF-κB signaling, was found as TANK-binding partners.[119] Therefore, the mechanism by which TANK inhibited TRAF6 ubiquitination has been elusive.
We recently reported that TANK represses genotoxic NF-κB activation, which may rely on suppression of TRAF6 ubiquitination.[121] TRAF6 polyubiquitination has been shown to play an important role in mediating IKK activation upon DNA damage.[70] DNA damage-induced NF-κB activation and TRAF6 ubiquitination were substantially increased in TANK-deficient cells which was reduced by reconstitution of TANK. TRAF6 was found to interact with TANK through its TRAF-C domain and this interaction is required for TANK-mediated deubiquitination of TRAF6. Intriguingly, TANK was identified as a MCPIP1-interacting protein in a proteomic screen and we confirmed that TANK forms a complex with MCPIP1 and USP10. The TANK-associated USP10 is required for the decrease of TRAF6 polyubiquitination, which in turn diminishes the NF-κB activation by DNA damage. Therefore, USP10 is able to efficiently terminate DNA damage-induced NF-κB activation through attenuating two critical ubiquitin events, linear ubiquitination of NEMO and K63-ubiquitination of TRAF6, in genotoxic NF-κB signaling. In addition to genotoxic stress, the TANK-MCPIP1-USP10 complex is also responsible for restraining TRAF6 ubiquitination in cells treated with LPS or IL-1β. Altogether, these data support that TANK may also serve as a negative regulator of genotoxic NF-κB activation by directing USP10-MCPIP1 complex to polyubiquitinated TRAF6.[121]
Inhibiting genotoxic NF-κB activation by targeting PARP-1
In cells undergoing apoptosis upon severe DNA damage, PARP-1 is cleaved by caspases which likely restrict any further activation of NF-κB. The PARP-1 cleavage not only diminishes the DNA repair capacity of the cells, but also blocks anti-apoptotic NF-κB activation in response to DNA damage, which ensures the elimination of cells with unrepairable DNA lesions. In signaling pathways leading to NF-κB activation, PARP-1 plays a unique role in mediating DNA damage-induced signaling cascade. DNA damage-induced NF-κB activation is believed to play important roles in mediating acquired resistance in cancer cells treated with genotoxic agents.[122,123] As NF-κB also has critical physiological functions, selective inhibition of NF-κB activated by genotoxic treatments is expected to effectively reduce therapeutic resistance to radiation and chemotherapies with minimal toxicity. Targeting PARP-1 may provide an attractive opportunity for selectively inhibiting genotoxic NF-κB activation while sparing the canonical and alternative NF-κB pathways.[124]
Physiological and pathological functions of genotoxic NF-κB activation
DNA damage-induced NF-κB activation and B-cell differentiation
Endogenous DSBs occurrence is an obligate consequence of B-lymphocyte development because of somatic rearrangement of immunoglobulin loci that is important for generation of antibody diversity and isotype class switching. These physiological DSBs mobilize the DDR machinery in a way reminiscent of the cell reaction to exogenous DNA damage.[125] As a result, mice deficient in DDR components, particularly in genes required for detecting and repair of DSBs (e.g. ATM), demonstrate defects in normal B-lymphocyte function and reactions to pathogens.[126] Also, in human, a number of essential immune deficiencies occur, owing to monogenic defects in DDR signaling.[127,128] Interestingly, NF-κB activation by endogenous DSBs formed during lymphocyte differentiation has been detected in mice.[129] Similar to the response to exogenous DSBs, NF-κB activation by endogenous DSBs requires NEMO and ATM signaling. In this scenario, NF-κB activation by endogenous DSBs up-regulates a cohort of genes including Pim2 and CD40, whose expression is required for preventing apoptosis and B-cell development. Consistently, patients with mutations in the gene encoding NEMO show defects in B-lymphocyte differentiation. Several mutations in NEMO map to the C-terminal ZF area that is vital for DNA damage-induced NF-κB activation. All tested NEMO ZF alleles have proven specifically defective for the genotoxic NF-κB pathway, although NF-κB activation by LPS stimulation remains largely intact. A prominent feature of B lymphocytes from patients with NEMO ZF mutations is the inability to carry out class switch recombination and almost complete absence of memory B cells.[130,131] Notably, microarray examination indicated that those genes required for class switch recombination and proliferation failed to be induced in patient B cells undergoing class switching in vitro.[131] This phenotype may be explained, to some degree, by the modest defects in CD40 signaling that are also observed in these samples. However, it is also speculated that the defect in B-cell functions observed in patients with NEMO ZF mutations may specifically require DNA damage-induced NF-κB activation.[132]
Constitutive ATM-NEMO-NF-κB activity in AML and MDS
Besides DNA damage-induced ATM-NEMO-dependent NF-κB activation, a recent study showed that ATM-NEMO-NF-κB signaling is constitutively activated in certain acute myeloid leukemia (AML) cell lines and in a high percentage of primary myelodysplastic syndrome (MDS) and AML patient samples.[133] Utilizing the P39 AML cell line, it was found that ATM is constitutively active with NEMO and PIDD in the nucleus, and an ATM-NEMO nuclear complex is clearly detectable without genotoxic treatments. Inhibition of ATM by KU55933 or ATM knockdown results in the loss of nuclear NEMO and PIDD, dissociation of ATM-NEMO complex, restraint of NF-κB activation, and induction of cell death. Also, active ATM was detected in CD34+ bone marrow mononuclear cells acquired from all high-risk MDS or AML patient samples, indicating constitutive ATM activation. In line with this notion, pharmacological inhibition of ATM induced cytoplasmic redistribution of NEMO and PIDD (which were found constitutively in the nucleus of these cells), inhibition of constitutive NF-κB activation and cell death.[133] Although constitutive activation of NF-κB is frequently found in various types of tumor specimens,[134-136] in many cases, the mechanisms that maintain NF-κB activity are unclear. Considering the genomic instability as a hallmark for human cancers,[137] it is tempting to speculate that endogenous DNA damage-induced signaling may account for, at least in part, maintaining constitutive NF-κB activation in various human malignancies, in addition to high-risk MDS and AML.
Genotoxic NF-κB activation in acquired cancer therapeutic resistance
Substantial evidence indicates that NF-κB regulates oncogenesis and tumor progression. NF-κB activation (i.e. nuclear localization) has been observed in a variety of solid tumors.[138,139] In general, NF-κB is believed to promote cancer development by contributing to all intrinsic hallmarks of cancer, including therapeutic resistance and metastasis.[137,138] Ionizing radiation and chemotherapeutic drugs such as doxorubicin, 5-fluorouracil and cisplatin have been found to activate NF-κB signaling in various cancer cells.[140-142] The genotoxic agent-induced NF-κB activation has been considered as a major mechanism through which various cancers acquire therapeutic resistance.[143]
A number of NF-κB target genes which prevent apoptosis and promote proliferation, such as cyclin D1, bcl-2, bcl-xL, survivin, and XIAP, were upregulated in cancer cells upon genotoxic treatments.[144-146] Furthermore, chemotherapeutic resistance of cancer cells highly correlates with their ability to metastasize, which indicates that genotoxic treatment may induce pro-metastatic responses in refractory cancer cells.[147]
Recent studies have suggested that DNA damage response could enhance the expression of pro-inflammatory cytokines, such as Interleukin-6 (IL-6) and Interleukin-8 (IL-8), favoring tumor growth, angiogenesis and malignant cell invasion.[148] Inflammation has been shown to play a significant role in promoting cancer metastasis.[149] As NF-κB is a key modulator of inflammation, it is plausible that chemotherapy-induced NF-κB activation may facilitate tumor metastasis by promoting an inflammatory response. Along with pro-inflammatory cytokines, accumulating evidence also suggests that certain cancer-related microRNAs may also play important roles in breast cancer metastasis.[150-152] MicroRNAs are a class of small non-coding RNAs (~20-24 nucleotides), which primarily bind to the 3′-untranslated region (3′-UTR) of target mRNA and negatively regulate gene expression at the post-transcriptional level.[153] Most miRNA genes are transcribed into primary miRNAs by RNA polymerase II, which can be sequentially processed into precursor, then mature miRNAs. The transcription of miRNA can be regulated by both upstream DNA transcription regulatory elements, such as conserved transcription factor binding sites, and epigenetic modifications.[154] Genotoxic stimulation was shown to alter miRNAs expression in cancer cells at transcriptional and post-transcriptional levels.[155-157] NF-κB has also emerged as an important transcription regulator of miRNA genes.[158-161] A recent report showed that genotoxic NF-κB activation regulates the expression of both pro-inflammatory cytokine IL-6 and oncogenic miR-21 in TNBC cells, which may promote TNBC cell survival and invasion upon Dox treatment.[162] Moreover, IL-6-dependent STAT3 activation further enhanced miR-181a transcription, whose upregulation suppressed pro-apoptotic gene BAX level and reduced cancer cell apoptosis upon chemotherapy.[163] It is plausible that NF-κB plays a critical role in regulating therapeutic resistance and subsequent metastasis in genotoxic drug-treated breast cancer cells, through coordinating expression of anti-apoptotic genes, pro-inflammatory cytokines and oncogenic miRNAs. Thus, inhibiting genotoxic drug-induced NF-κB activation may serve as a promising strategy to reduce chemotherapeutic resistance and subsequent metastasis.
Targeting NF-κB to suppress therapeutic resistance
A number of NF-κB blocking agents, such as IKK inhibitors, inhibitory peptides, antisense RNA, proteasome inhibitors and dietary supplements, are currently being tested in combination with chemotherapy and radiotherapy. These studies aim to sensitize cancer cells to the tumoricidal effects of chemotherapeutic drugs and radiation by blocking NF-κB activation, thereby preventing acquired resistance. It is noteworthy that NF-κB also plays critical roles in regulating physiological process, such as immune response. Non-selectively inhibiting NF-κB may lead to severe adverse effects such as immunodeficiency. Whether such a NF-κB inhibitor will do more harm than good with respect to the immune system in cancer patients is likely to depend on the particular target or combination of targets on which it acts.
In this regard, agents selectively targeting NF-κB signaling activated specifically by DNA damage are expected to show much greater promise in antagonizing therapeutic resistance without compromising the immune system. The recent development of PARP-1 inhibitors may provide an extraordinary opportunity for clinical application of such a genotoxic NF-κB selective inhibitor. It has been shown that PARP inhibitors significantly augmented cell death in BRCA1/2-deficient cancer cells, while no overt toxicity was observed in normal cells expressing functional BRCA1/2.[164,165] This effect was termed “synthetic lethality”, which is believed to be a promising “targeted” strategy to selectively eliminate cancer cells harboring BRCA1/2 mutation while sparing normal cells.[166,167] Currently, FDA has approved the PARP inhibitors olaparib and rucaparib for treating ovarian cancer patients with BRCA mutation. It has been shown that DNA damage-induced NF-κB activation in human cancer cells was significantly attenuated by PARP-1 inhibitors.[168] Moreover, a PARP1 inhibitor, AG-014699 was shown to sensitize glioma cells to radiation via inhibiting NF-κB activation.[169] Thus, inhibiting PARP1 may not only further diminish the DNA repair capacity of BRCA-deficient breast cancer cells, but also abolish genotoxic drug-induced NF-κB activation and sensitize TNBC patients to chemotherapy.
Conclusion and perspective
The studies on DNA damage response and NF-κB have significantly improved our understanding of molecular signaling leading to genotoxic NF-κB activation in the last two decades. These studies have also provided promising drug targets, such as PARP-1, for selectively inhibiting NF-κB activation by radiation and chemotherapeutics in cancer cells, which may be able to resensitize treatment-refractory cancer cells to conventional chemotherapy. The extension of the period that cancer patients can benefit from these cost-effective “old” chemotherapeutic drugs will also substantially alleviate the financial burden the patients bear due to the high cost of newly developed therapeutic agents. Nevertheless, further investigation is still much needed to explore the critical downstream NF-κB-target genes which are specifically induced by DNA damage. Better understanding of how these induced genes, protein genes or non-coding RNAs, modulate cell response to DNA damage, will help to develop novel therapeutic agents selectively targeting those pro-survival/metastatic targets and counteract acquired therapeutic resistance. Furthermore, these gene signatures may also serve as predictive biomarkers for evaluating the potential benefit and effectiveness in patients who receive cytotoxic chemotherapies and radiation.
Authors’ contributions
The study’s conception and design: W. Wang, A.M. Mani, Z.H. Wu
Paper’s writing: W. Wang, A.M. Mani, Z.H. Wu
Manuscripts review and edition: Z.H. Wu
Financial support and sponsorship
The work in the authors’ laboratory has been supported by NIH R01CA149251 and American Cancer Society (RSG-13-186-01-CSM).
Conflicts of interest
There are no conflicts of interest.
Patient consent
There is no patient involved.
Ethics approval
This article does not contain any studies with human participants or animals.
REFERENCES
1. Sancar A, Lindsey-Boltz LA, Unsal-Kacmaz K, Linn S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu Rev Biochem 2004;73:39-85.
2. Khanna KK, Jackson SP. DNA double-strand breaks: signaling, repair and the cancer connection. Nat Genet 2001;27:247-54.
3. De Bont R, van Larebeke N. Endogenous DNA damage in humans: a review of quantitative data. Mutagenesis 2004;19:169-85.
4. Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol Cell 2010;40:179-204.
5. d'Adda di Fagagna F. Living on a break: cellular senescence as a DNA-damage response. Nat Rev Cancer 2008;8:512-22.
6. McCool KW, Miyamoto S. DNA damage-dependent NF-kappa B activation: NEMO turns nuclear signaling inside out. Immunol Rev 2012;246:311-26.
7. Sen R, Baltimore D. Inducibility of kappa immunoglobulin enhancer-binding protein Nf-kappa B by a posttranslational mechanism. Cell 1986;47:921-8.
8. Sen R, Baltimore D. Multiple nuclear factors interact with the immunoglobulin enhancer sequences. Cell 1986.46:705-716. J Immunol 2006;177:7485-96.
9. Baeuerle PA, Baltimore D. I kappa B: a specific inhibitor of the NF-kappa B transcription factor. Science 1988;242:540-6.
12. Piret B, Piette J. Topoisomerase poisons activate the transcription factor NF-kappaB in ACH-2 and CEM cells. Nucleic Acids Res 1996;24:4242-8.
13. Habraken Y, Piret B, Piette J. S phase dependence and involvement of NF-kappaB activating kinase to NF-kappaB activation by camptothecin. Biochem Pharmacol 2001;62:603-16.
14. Wu ZH, Miyamoto S. Many faces of NF-kappa B signaling induced by genotoxic stress. J Mol Med (Berl) 2007;85:1187-202.
15. Schmitt AM, Crawley CD, Kang S, Raleigh DR, Yu X, Wahlstrom JS, Voce DJ, Darga TE, Weichselbaum RR, Yamini B. p50 (NF-kappaB1) is an effector protein in the cytotoxic response to DNA methylation damage. Mol Cell 2011;44:785-96.
16. Campbell KJ, Witty JM, Rocha S, Perkins ND. Cisplatin mimics ARF tumor suppressor regulation of RelA (p65) nuclear factor-kappaB transactivation. Cancer Res 2006;66:929-35.
17. Hartlerode AJ, Scully R. Mechanisms of double-strand break repair in somatic mammalian cells. Biochem J 2009;423:157-68.
18. Goodarzi AA, Noon AT, Jeggo PA. The impact of heterochromatin on DSB repair. Biochem Soc Trans 2009;37:569-76.
19. Brown JS, O'Carrigan B, Jackson SP, Yap TA. Targeting DNA repair in cancer: beyond PARP inhibitors. Cancer Discov 2017;7:20-37.
20. Vilenchik MM, Knudson AG. Endogenous DNA double-strand breaks: production, fidelity of repair, and induction of cancer. Proc Natl Acad Sci U S A 2003;100:12871-6.
21. Branzei D, Foiani M. Maintaining genome stability at the replication fork. Nat Rev Mol Cell Biol 2010;11:208-19.
22. Dillon LW, Burrow AA, Wang YH. DNA instability at chromosomal fragile sites in cancer. Curr Genomics 2010;11:326-37.
23. Stracker TH, Theunissen JW, Morales M, Petrini JH. The Mre11 complex and the metabolism of chromosome breaks: the importance of communicating and holding things together. DNA Repair (Amst) 2004;3:845-54.
24. Lavin MF. ATM and the Mre11 complex combine to recognize and signal DNA double-strand breaks. Oncogene 2007;26:7749-58.
25. Lee JH, Paull TT. Activation and regulation of ATM kinase activity in response to DNA double-strand breaks. Oncogene 2007;26:7741-8.
26. Sun Y, Jiang X, Chen S, Fernandes N, Price BD. A role for the Tip60 histone acetyltransferase in the acetylation and activation of ATM. Proc Natl Acad Sci U S A 2005;102:13182-7.
27. Goodarzi AA, Jonnalagadda JC, Douglas P, Young D, Ye R, Moorhead GB, Lees-Miller SP, Khanna KK. Autophosphorylation of ataxia-telangiectasia mutated is regulated by protein phosphatase 2A. EMBO J 2004;23:4451-61.
28. Sun Y, Jiang X, Xu Y, Ayrapetov MK, Moreau LA, Whetstine JR, Price BD. Histone H3 methylation links DNA damage detection to activation of the tumour suppressor Tip60. Nat Cell Biol 2009;11:1376-82.
29. Sun Y, Xu Y, Roy K, Price BD. DNA damage-induced acetylation of lysine 3016 of ATM activates ATM kinase activity. Mol Cell Biol 2007;27:8502-9.
30. Bakkenist CJ, Kastan MB. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature 2003;421:499-506.
31. Kozlov SV, Graham ME, Jakob B, Tobias F, Kijas AW, Tanuji M, Chen P, Robinson PJ, Taucher-Scholz G, Suzuki K, So S, Chen D, Lavin MF. Autophosphorylation and ATM activation: additional sites add to the complexity. J Biol Chem 2011;286:9107-19.
32. Kozlov SV, Graham ME, Peng C, Chen P, Robinson PJ, Lavin MF. Involvement of novel autophosphorylation sites in ATM activation. EMBO J 2006;25:3504-14.
33. Polo SE, Jackson SP. Dynamics of DNA damage response proteins at DNA breaks: a focus on protein modifications. Genes Dev 2011;25:409-33.
34. Andegeko Y, Moyal L, Mittelman L, Tsarfaty I, Shiloh Y, Rotman G. Nuclear retention of ATM at sites of DNA double strand breaks. J Biol Chem 2001;276:38224-30.
35. Matsuoka S, Ballif BA, Smogorzewska A, McDonald ER 3rd, Hurov KE, Luo J, Bakalarski CE, Zhao Z, Solimini N, Lerenthal Y, Shiloh Y, Gygi SP, Elledge SJ. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 2007;316:1160-6.
36. Neal JA, Meek K. Choosing the right path: does DNA-PK help make the decision? Mutat Res 2011;711:73-86.
37. Burma S, Chen DJ. Role of DNA-PK in the cellular response to DNA double-strand breaks. DNA Repair (Amst) 2004;3:909-18.
38. Jiang X, Sun Y, Chen S, Roy K, Price BD. The FATC domains of PIKK proteins are functionally equivalent and participate in the Tip60-dependent activation of DNA-PKcs and ATM. J Biol Chem 2006;281:15741-6.
39. Hill R, Lee PW. The DNA-dependent protein kinase (DNA-PK): more than just a case of making ends meet? Cell Cycle 2010;9:3460-9.
40. Stiff T, O'Driscoll M, Rief N, Iwabuchi K, Lobrich M, Jeggo PA. ATM and DNA-PK function redundantly to phosphorylate H2AX after exposure to ionizing radiation. Cancer Res 2004;64:2390-6.
41. Flynn RL, Zou L. ATR: a master conductor of cellular responses to DNA replication stress. Trends Biochem Sci 2011;36:133-40.
42. Yan S, Michael WM. TopBP1 and DNA polymerase alpha-mediated recruitment of the 9-1-1 complex to stalled replication forks: implications for a replication restart-based mechanism for ATR checkpoint activation. Cell Cycle 2009;8:2877-84.
43. Paulsen RD, Cimprich KA. The ATR pathway: fine-tuning the fork. DNA Repair (Amst) 2007;6:953-66.
44. Brown EJ, Baltimore D. Essential and dispensable roles of ATR in cell cycle arrest and genome maintenance. Genes Dev 2003;17:615-28.
45. de Klein A, Muijtjens M, van Os R, Verhoeven Y, Smit B, Carr AM, Lehmann AR, Hoeijmakers JH. Targeted disruption of the cell-cycle checkpoint gene ATR leads to early embryonic lethality in mice. Curr Biol 2000;10:479-82.
46. Elkon R, Rashi-Elkeles S, Lerenthal Y, Linhart C, Tenne T, Amariglio N, Rechavi G, Shamir R, Shiloh Y. Dissection of a DNA-damage-induced transcriptional network using a combination of microarrays, RNA interference and computational promoter analysis. Genome Biol 2005;6:R43.
47. Rashi-Elkeles S, Elkon R, Weizman N, Linhart C, Amariglio N, Sternberg G, Rechavi G, Barzilai A, Shamir R, Shiloh Y. Parallel induction of ATM-dependent pro- and antiapoptotic signals in response to ionizing radiation in murine lymphoid tissue. Oncogene 2006;25:1584-92.
48. Rashi-Elkeles S, Warnatz HJ, Elkon R, Kupershtein A, Chobod Y, Paz A, Amstislavskiy V, Sultan M, Safer H, Nietfeld W, Lehrach H, Shamir R, Yaspo ML, Shiloh Y. Parallel profiling of the transcriptome, cistrome, and epigenome in the cellular response to ionizing radiation. Sci Signal 2014;7:rs3.
49. Shiloh Y, Ziv Y. The ATM protein kinase: regulating the cellular response to genotoxic stress, and more. Nat Rev Mol Cell Biol 2013;14:197-210.
50. Goldstein M, Kastan MB. The DNA damage response: implications for tumor responses to radiation and chemotherapy. Annu Rev Med 2015;66:129-43.
51. Ghosh S, May MJ, Kopp EB. NF-kappa B and Rel proteins: evolutionarily conserved mediators of immune responses. Annu Rev Immunol 1998;16:225-60.
52. Perkins ND, Gilmore TD. Good cop, bad cop: the different faces of NF-kappa B. Cell Death Differ 2006;13:759-72.
53. Vallabhapurapu S, Karin M. Regulation and function of NF-kappaB transcription factors in the immune system. Annu Rev Immuno 2009;27:693-733.
55. Lee SJ, Dimtchev A, Lavin MF, Dritschilo A, Jung M. A novel ionizing radiation-induced signaling pathway that activates the transcription factor NF-kappaB. Oncogene 1998;17:1821-6.
56. Piret B, Schoonbroodt S, Piette J. The ATM protein is required for sustained activation of NF-kappaB following DNA damage. Oncogene 1999;18:2261-71.
57. Hwang B, McCool K, Wan J, Wuerzberger-Davis SM, Young EW, Choi EY, Cingolani G, Weaver BA, Miyamoto S. IPO3-mediated nonclassical nuclear import of NF-κB essential modulator (NEMO) drives DNA damage-dependent NF-κB activation. J Biol Chem 2015;290:17967-84.
58. Huang TT, Wuerzberger-Davis SM, Wu ZH, Miyamoto S. Sequential modification of NEMO/IKKgamma by SUMO-1 and ubiquitin mediates NF-kappaB activation by genotoxic stress. Cell 2003;115:565-76.
59. Mabb AM, Wuerzberger-Davis SM, Miyamoto S. PIASy mediates NEMO sumoylation and NF-kappaB activation in response to genotoxic stress. Nat Cell Biol 2006;8:986-93.
60. Janssens S, Tinel A, Lippens S, Tschopp J. PIDD mediates NF-kappaB activation in response to DNA damage. Cell 2005;123:1079-92.
61. Krishnakumar R, Kraus WL. The PARP side of the nucleus: molecular actions, physiological outcomes, and clinical targets. Mol Cell 2010;39:8-24.
62. Rouleau M, Patel A, Hendzel MJ, Kaufmann SH, Poirier GG. PARP inhibition: PARP1 and beyond. Nat Rev Cancer 2010;10:293-301.
63. Malanga M, Althaus FR. The role of poly(ADP-ribose) in the DNA damage signaling network. Biochem Cell Biol 2005;83:354-64.
64. Leppard JB, Dong Z, Mackey ZB, Tomkinson AE. Physical and functional interaction between DNA ligase IIIalpha and poly(ADP-Ribose) polymerase 1 in DNA single-strand break repair. Mol Cell Biol 2003;23:5919-27.
65. Stilmann M, Hinz M, Arslan SC, Zimmer A, Schreiber V, Scheidereit C. A nuclear poly(ADP-ribose)-dependent signalosome confers DNA damage-induced IkappaB kinase activation. Mol Cell 2009;36:365-78.
66. Fu K, Sun X, Wier EM, Hodgson A, Liu Y, Sears CL, Wan F. Sam68/KHDRBS1 is critical for colon tumorigenesis by regulating genotoxic stress-induced NF-kappaB activation. Elife 2016;5:e15018.
67. Sheng YH, He Y, Hasnain SZ, Wang R, Tong H, Clarke DT, Lourie R, Oancea I, Wong KY, Lumley JW, Florin TH, Sutton P, Hooper JD, McMillan NA, McGuckin MA. MUC13 protects colorectal cancer cells from death by activating the NF-kappaB pathway and is a potential therapeutic target. Oncogene 2017;36:700-13.
68. Wu ZH, Shi Y, Tibbetts RS, Miyamoto S. Molecular linkage between the kinase ATM and NF-kappaB signaling in response to genotoxic stimuli. Science 2006;311:1141-6.
69. Jin HS, Lee DH, Kim DH, Chung JH, Lee SJ, Lee TH. cIAP1, cIAP2, and XIAP act cooperatively via nonredundant pathways to regulate genotoxic stress-induced nuclear factor-kappaB activation. Cancer Res 2009;69:1782-91.
70. Hinz M, Stilmann M, Arslan SC, Khanna KK, Dittmar G, Scheidereit C. A cytoplasmic ATM-TRAF6-cIAP1 module links nuclear DNA damage signaling to ubiquitin-mediated NF-kappaB activation. Mol Cell 2010;40:63-74.
71. Schoch S, Gundelfinger E. Molecular organization of the presynaptic active zone. Cell Tissue Res 2006;326:379-91.
72. Ohtsuka T, Takao-Rikitsu E, Inoue E, Inoue M, Takeuchi M, Matsubara K, Deguchi-Tawarada M, Satoh K, Morimoto K, Nakanishi H, Takai Y. Cast: a novel protein of the cytomatrix at the active zone of synapses that forms a ternary complex with RIM1 and munc13-1. J Cell Biol 2002;158:577-90.
73. Wang Y, Liu X, Biederer T, Sudhof TC. A family of RIM-binding proteins regulated by alternative splicing: implications for the genesis of synaptic active zones. Proc Natl Acad Sci U S A 2002;99:14464-9.
74. Ducut-Sigala JL, Bottero V, Young DB, Shevchenko A, Mercurio F, Verma IM. Activation of transcription factor NF-kappaB requires ELKS, an IkappaB kinase regulatory subunit. Science 2004;304:1963-7.
75. Wu ZH, Wong ET, Shi Y, Niu J, Chen Z, Miyamoto S, Tergaonkar V. ATM- and NEMO-dependent ELKS ubiquitination coordinates TAK1-mediated IKK activation in response to genotoxic stress. Mol Cell 2010;40:75-86.
76. Niu J, Shi Y, Iwai K, Wu ZH. LUBAC regulates NF-kappaB activation upon genotoxic stress by promoting linear ubiquitination of NEMO. EMBO J 2011;30:3741-53.
77. Komander D, Reyes-Turcu F, Licchesi JDF, Odenwaelder P, Wilkinson KD, Barford D. Molecular discrimination of structurally equivalent Lys 63-linked and linear polyubiquitin chains. EMBO Rep 2009;10:466-73.
78. Kirisako T, Kamei K, Murata S, Kato M, Fukumoto H, Kanie M, Sano S, Tokunaga F, Tanaka K, Iwai K. A ubiquitin ligase complex assembles linear polyubiquitin chains. Embo J 2006;25:4877-87.
79. Gerlach B, Cordier SM, Schmukle AC, Emmerich CH, Rieser E, Haas TL, Webb AI, Rickard JA, Anderton H, Wong WWL, Nachbur U, Gangoda L, Warnken U, Purcell AW, Silke J, Walczak H. Linear ubiquitination prevents inflammation and regulates immune signalling. Nature 2011;471:591-6.
80. Ikeda F, Deribe YL, Skanland SS, Stieglitz B, Grabbe C, Franz-Wachtel M, van Wijk SJL, Goswami P, Nagy V, Terzic J, Tokunaga F, Androulidaki A, Nakagawa T, Pasparakis M, Iwai K, Sundberg JP, Schaefer L, Rittinger K, Macek B, Dikic I. SHARPIN forms a linear ubiquitin ligase complex regulating NF-κB activity and apoptosis. Nature 2011;471:637-41.
81. Tokunaga F, Nakagawa T, Nakahara M, Saeki Y, Taniguchi M, Sakata S, Tanaka K, Nakano H, Iwai K. SHARPIN is a component of the NF-kB-activating linear ubiquitin chain assembly complex. Nature 2011;471:633-6.
82. Haas TL, Emmerich CH, Gerlach B, Schmukle AC, Cordier SM, Rieser E, Feltham R, Vince J, Warnken U, Wenger T, Koschny R, Komander D, Silke J, Walczak H. Recruitment of the linear ubiquitin chain assembly complex stabilizes the TNF-R1 signaling complex and is required for TNF-mediated gene induction. Mol Cell 2009;36:831-44.
83. Tokunaga F, Sakata S, Saeki Y, Satomi Y, Kirisako T, Kamei K, Nakagawa T, Kato M, Murata S, Yamaoka S, Yamamoto M, Akira S, Takao T, Tanaka K, Iwai K. Involvement of linear polyubiquitylation of NEMO in NF-kappaB activation. Nat Cell Biol 2009;11:123-32.
84. Lo YC, Lin SC, Rospigliosi CC, Conze DB, Wu CJ, Ashwell JD, Eliezer D, Wu H. Structural basis for recognition of diubiquitins by NEMO. Mol Cell 2009;33:602-15.
85. Rahighi S, Ikeda F, Kawasaki M, Akutsu M, Suzuki N, Kato R, Kensche T, Uejima T, Bloor S, Komander D, Randow F, Wakatsuki S, Dikic I. Specific recognition of linear ubiquitin chains by NEMO is important for NF-kappaB activation. Cell 2009;136:1098-109.
86. Josson S, Xu Y, Fang F, Dhar SK, St Clair DK, St Clair WH. RelB regulates manganese superoxide dismutase gene and resistance to ionizing radiation of prostate cancer cells. Oncogene 2006;25:1554-9.
87. Xu Y, Fang F, St Clair DK, Sompol P, Josson S, St Clair WH. SN52, a novel nuclear factor-kappaB inhibitor, blocks nuclear import of RelB:p52 dimer and sensitizes prostate cancer cells to ionizing radiation. Mol Cancer Ther 2008;7:2367-76.
88. Lessard L, Begin LR, Gleave ME, Mes-Masson AM, Saad F. Nuclear localisation of nuclear factor-kappaB transcription factors in prostate cancer: an immunohistochemical study. Br J Cancer 2005;93:1019-23.
89. Renner F, Schmitz ML. Autoregulatory feedback loops terminating the NF-kappaB response. Trends Biochem Sci 2009;34:128-35.
91. Chiao PJ, Miyamoto S, Verma IM. Autoregulation of I kappa B alpha activity. Proc Natl Acad Sci U S A 1994;91:28-32.
92. Sun SC, Ganchi PA, Ballard DW, Greene WC. NF-kappa B controls expression of inhibitor I kappa B alpha: evidence for an inducible autoregulatory pathway. Science 1993;259:1912-5.
93. Beg AA, Sha WC, Bronson RT, Baltimore D. Constitutive NF-kappa B activation, enhanced granulopoiesis, and neonatal lethality in I kappa B alpha-deficient mice. Genes Dev 1995;9:2736-46.
94. Brummelkamp TR, Nijman SM, Dirac AM, Bernards R. Loss of the cylindromatosis tumour suppressor inhibits apoptosis by activating NF-kappaB. Nature 2003;424:797-801.
95. Jono H, Lim JH, Chen LF, Xu H, Trompouki E, Pan ZK, Mosialos G, Li JD. NF-kappaB is essential for induction of CYLD, the negative regulator of NF-kappaB: evidence for a novel inducible autoregulatory feedback pathway. J Biol Chem 2004;279:36171-4.
96. Wertz IE, O'Rourke KM, Zhou H, Eby M, Aravind L, Seshagiri S, Wu P, Wiesmann C, Baker R, Boone DL, Ma A, Koonin EV, Dixit VM. De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-kappaB signalling. Nature 2004;430:694-9.
97. Lee EG, Boone DL, Chai S, Libby SL, Chien M, Lodolce JP, Ma A. Failure to regulate TNF-induced NF-kappaB and cell death responses in A20-deficient mice. Science 2000;289:2350-4.
98. Cheng J, Kang X, Zhang S, Yeh ET. SUMO-specific protease 1 is essential for stabilization of HIF1alpha during hypoxia. Cell 2007;131:584-95.
100. Lee MH, Mabb AM, Gill GB, Yeh ET, Miyamoto S. NF-kappaB induction of the SUMO protease SENP2: a negative feedback loop to attenuate cell survival response to genotoxic stress. Mol Cell 2011;43:180-91.
101. Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, Wei G, Chepelev I, Zhao K. High-resolution profiling of histone methylations in the human genome. Cell 2007;129:823-37.
102. Metzger E, Imhof A, Patel D, Kahl P, Hoffmeyer K, Friedrichs N, Muller JM, Greschik H, Kirfel J, Ji S, Kunowska N, Beisenherz-Huss C, Gunther T, Buettner R, Schule R. Phosphorylation of histone H3T6 by PKCbeta(I) controls demethylation at histone H3K4. Nature 2010;464:792-6.
103. Kanoh J, Francesconi S, Collura A, Schramke V, Ishikawa F, Baldacci G, Geli V. The fission yeast spSet1p is a histone H3-K4 methyltransferase that functions in telomere maintenance and DNA repair in an ATM kinase Rad3-dependent pathway. J Mol Biol 2003;326:1081-94.
104. Moyal L, Lerenthal Y, Gana-Weisz M, Mass G, So S, Wang SY, Eppink B, Chung YM, Shalev G, Shema E, Shkedy D, Smorodinsky NI, van Vliet N, Kuster B, Mann M, Ciechanover A, Dahm-Daphi J, Kanaar R, Hu MC, Chen DJ, Oren M, Shiloh Y. Requirement of ATM-dependent monoubiquitylation of histone H2B for timely repair of DNA double-strand breaks. Mol Cell 2011;41:529-42.
105. Nakamura K, Kato A, Kobayashi J, Yanagihara H, Sakamoto S, Oliveira DV, Shimada M, Tauchi H, Suzuki H, Tashiro S, Zou L, Komatsu K. Regulation of homologous recombination by RNF20-dependent H2B ubiquitination. Mol Cell 2011;41:515-28.
106. Sun SC. CYLD: a tumor suppressor deubiquitinase regulating NF-kappaB activation and diverse biological processes. Cell Death Differ 2010;17:25-34.
107. Zhou L, Azfer A, Niu J, Graham S, Choudhury M, Adamski FM, Younce C, Binkley PF, Kolattukudy PE. Monocyte chemoattractant protein-1 induces a novel transcription factor that causes cardiac myocyte apoptosis and ventricular dysfunction. Circ Res 2006;98:1177-85.
108. Liang J, Wang J, Azfer A, Song W, Tromp G, Kolattukudy PE, Fu M. A novel CCCH-zinc finger protein family regulates proinflammatory activation of macrophages. J Biol Chem 2008;283:6337-46.
109. Liang J, Saad Y, Lei T, Wang J, Qi D, Yang Q, Kolattukudy PE, Fu M. MCP-induced protein 1 deubiquitinates TRAF proteins and negatively regulates JNK and NF-kappaB signaling. J Exp Med 2010;207:2959-73.
110. Matsushita K, Takeuchi O, Standley DM, Kumagai Y, Kawagoe T, Miyake T, Satoh T, Kato H, Tsujimura T, Nakamura H, Akira S. Zc3h12a is an RNase essential for controlling immune responses by regulating mRNA decay. Nature 2009;458:1185-90.
111. Skalniak L, Mizgalska D, Zarebski A, Wyrzykowska P, Koj A, Jura J. Regulatory feedback loop between NF-kappaB and MCP-1-induced protein 1 RNase. FEBS J 2009;276:5892-905.
112. Niu J, Shi Y, Xue J, Miao R, Huang S, Wang T, Wu J, Fu M, Wu ZH. USP10 inhibits genotoxic NF-kappaB activation by MCPIP1-facilitated deubiquitination of NEMO. EMBO J 2013;32:3206-19.
113. Pomerantz JL, Baltimore D. NF-kappaB activation by a signaling complex containing TRAF2, TANK and TBK1, a novel IKK-related kinase. EMBO J 1999;18:6694-704.
114. Rothe M, Xiong J, Shu HB, Williamson K, Goddard A, Goeddel DV. I-TRAF is a novel TRAF-interacting protein that regulates TRAF-mediated signal transduction. Proc Natl Acad Sci U S A 1996;93:8241-6.
115. Cheng G, Baltimore D. TANK, a co-inducer with TRAF2 of TNF- and CD 40L-mediated NF-kappaB activation. Genes Dev 1996;10:963-73.
116. Sharma S, tenOever BR, Grandvaux N, Zhou GP, Lin R, Hiscott J. Triggering the interferon antiviral response through an IKK-related pathway. Science 2003;300:1148-51.
117. Hacker H, Redecke V, Blagoev B, Kratchmarova I, Hsu LC, Wang GG, Kamps MP, Raz E, Wagner H, Hacker G, Mann M, Karin M. Specificity in Toll-like receptor signalling through distinct effector functions of TRAF3 and TRAF6. Nature 2006;439:204-7.
118. Oganesyan G, Saha SK, Guo B, He JQ, Shahangian A, Zarnegar B, Perry A, Cheng G. Critical role of TRAF3 in the Toll-like receptor-dependent and -independent antiviral response. Nature 2006;439:208-11.
119. Kawagoe T, Takeuchi O, Takabatake Y, Kato H, Isaka Y, Tsujimura T, Akira S. TANK is a negative regulator of Toll-like receptor signaling and is critical for the prevention of autoimmune nephritis. Nat immunol 2009;10:965-72.
120. Maruyama K, Kawagoe T, Kondo T, Akira S, Takeuchi O. TRAF family member-associated NF-kappaB activator (TANK) is a negative regulator of osteoclastogenesis and bone formation. J Biol Chem 2012;287:29114-24.
121. Wang W, Huang X, Xin HB, Fu M, Xue A, Wu ZH. TRAF Family Member-associated NF-kappaB Activator (TANK) Inhibits Genotoxic Nuclear Factor kappaB Activation by Facilitating Deubiquitinase USP10-dependent Deubiquitination of TRAF6 Ligase. J Biol Chem 2015;290:13372-85.
122. Baldwin AS. Regulation of cell death and autophagy by IKK and NF-kappaB: critical mechanisms in immune function and cancer. Immunol Rev 2012;246:327-45.
123. Orlowski RZ, Baldwin AS. NF-kappaB as a therapeutic target in cancer. Trends Mol Med 2002;8:385-9.
124. Veuger SJ, Hunter JE, Durkacz BW. Ionizing radiation-induced NF-kappaB activation requires PARP-1 function to confer radioresistance. Oncogene 2009;28:832-42.
125. Bassing CH, Alt FW. The cellular response to general and programmed DNA double strand breaks. DNA Repair (Amst) 2004;3:781-96.
126. Lumsden JM, McCarty T, Petiniot LK, Shen R, Barlow C, Wynn TA, Morse HC 3rd, Gearhart PJ, Wynshaw-Boris A, Max EE, Hodes RJ. Immunoglobulin class switch recombination is impaired in Atm-deficient mice. J Exp Med 2004;200:1111-21.
127. Slatter MA, Gennery AR. Primary immunodeficiencies associated with DNA-repair disorders. Exp Rev Mol Med 2010;12:e9.
128. Lavin MF. Ataxia-telangiectasia: from a rare disorder to a paradigm for cell signalling and cancer. Nat Rev Mol Cell Biol 2008;9:759-69.
129. Bredemeyer AL, Helmink BA, Innes CL, Calderon B, McGinnis LM, Mahowald GK, Gapud EJ, Walker LM, Collins JB, Weaver BK, Mandik-Nayak L, Schreiber RD, Allen PM, May MJ, Paules RS, Bassing CH, Sleckman BP. DNA double-strand breaks activate a multi-functional genetic program in developing lymphocytes. Nature 2008;456:819-23.
130. Jain A, Ma CA, Liu S, Brown M, Cohen J, Strober W. Specific missense mutations in NEMO result in hyper-IgM syndrome with hypohydrotic ectodermal dysplasia. Nat Immunol 2001;2:223-8.
131. Jain A, Ma CA, Lopez-Granados E, Means G, Brady W, Orange JS, Liu S, Holland S, Derry JM. Specific NEMO mutations impair CD40-mediated c-Rel activation and B cell terminal differentiation. J Clin Invest 2004;114:1593-602.
132. Huang TT, Feinberg SL, Suryanarayanan S, Miyamoto S. The zinc finger domain of NEMO is selectively required for NF-kappa B activation by UV radiation and topoisomerase inhibitors. Mol Cell Biol 2002;22:5813-25.
133. Grosjean-Raillard J, Tailler M, Ades L, Perfettini JL, Fabre C, Braun T, De Botton S, Fenaux P, Kroemer G. ATM mediates constitutive NF-kappaB activation in high-risk myelodysplastic syndrome and acute myeloid leukemia. Oncogene 2009;28:1099-109.
134. Keats JJ, Fonseca R, Chesi M, Schop R, Baker A, Chng WJ, Van Wier S, Tiedemann R, Shi CX, Sebag M, Braggio E, Henry T, Zhu YX, Fogle H, Price-Troska T, Ahmann G, Mancini C, Brents LA, Kumar S, Greipp P, Dispenzieri A, Bryant B, Mulligan G, Bruhn L, Barrett M, Valdez R, Trent J, Stewart AK, Carpten J, Bergsagel PL. Promiscuous mutations activate the noncanonical NF-kappaB pathway in multiple myeloma. Cancer Cell 2007;12:131-44.
135. Meylan E, Dooley AL, Feldser DM, Shen L, Turk E, Ouyang C, Jacks T. Requirement for NF-kappaB signalling in a mouse model of lung adenocarcinoma. Nature 2009;462:104-7.
136. Boehm JS, Zhao JJ, Yao J, Kim SY, Firestein R, Dunn IF, Sjostrom SK, Garraway LA, Weremowicz S, Richardson AL, Greulich H, Stewart CJ, Mulvey LA, Shen RR, Ambrogio L, Hirozane-Kishikawa T, Hill DE, Vidal M, Meyerson M, Grenier JK, Hinkle G, Root DE, Roberts TM, Lander ES, Polyak K, Hahn WC. Integrative genomic approaches identify IKBKE as a breast cancer oncogene. Cell 2007;129:1065-79.
139. Kim HJ, Hawke N, Baldwin AS. NF-kappaB and IKK as therapeutic targets in cancer. Cell Death Differ 2006;13:738-47.
140. Melisi D, Chiao PJ. NF-kappa B as a target for cancer therapy. Expert Opin Ther Targets 2007;11:133-44.
141. Baumann P, Mandl-Weber S, Oduncu F, Schmidmaier R. Alkylating agents induce activation of NFkappaB in multiple myeloma cells. Leuk Res 2008;32:1144-7.
142. Bednarski BK, Ding X, Coombe K, Baldwin AS, Kim HJ. Active roles for inhibitory kappaB kinases alpha and beta in nuclear factor-kappaB-mediated chemoresistance to doxorubicin. Mol Cancer Ther 2008;7:1827-35.
143. Baldwin AS. Control of oncogenesis and cancer therapy resistance by the transcription factor NF-kappaB. J Clin Invest 2001;107:241-6.
144. Tamatani M, Che YH, Matsuzaki H, Ogawa S, Okado H, Miyake S, Mizuno T, Tohyama M. Tumor necrosis factor induces Bcl-2 and Bcl-x expression through NFkappaB activation in primary hippocampal neurons. J Biol Chem 1999;274:8531-8.
145. Guttridge DC, Albanese C, Reuther JY, Pestell RG, Baldwin AS Jr. NF-kappaB controls cell growth and differentiation through transcriptional regulation of cyclin D1. Mol Cell Biol 1999;19:5785-99.
146. Stehlik C, de Martin R, Kumabashiri I, Schmid JA, Binder BR, Lipp J. Nuclear factor (NF)-kappaB-regulated X-chromosome-linked iap gene expression protects endothelial cells from tumor necrosis factor alpha-induced apoptosis. J Exp Med 1998;188:211-6.
147. DeNardo DG, Brennan DJ, Rexhepaj E, Ruffell B, Shiao SL, Madden SF, Gallagher WM, Wadhwani N, Keil SD, Junaid SA, Rugo HS, Hwang ES, Jirström K, West BL, Coussens LM. Leukocyte complexity predicts breast cancer survival and functionally regulates response to chemotherapy. Cancer Discov 2011;1:54-67.
148. Naugler WE, Karin M. The wolf in sheep's clothing: the role of interleukin-6 in immunity, inflammation and cancer. Trends Mol Med 2008;14:109-19.
150. White NMA, Fatoohi E, Metias M, Jung K, Stephan C, Yousef GM. Metastamirs: a stepping stone towards improved cancer management. Nat Rev Clin Oncol 2011;8:75-84.
151. Volinia S, Galasso M, Sana ME, Wise TF, Palatini J, Huebner K, Croce CM. Breast cancer signatures for invasiveness and prognosis defined by deep sequencing of microRNA. Proc Natl Acad Sci U S A 2012;109:3024-9.
154. Ozsolak F, Poling LL, Wang Z, Liu H, Liu XS, Roeder RG, Zhang X, Song JS, Fisher DE. Chromatin structure analyses identify miRNA promoters. Genes Dev 2008;22:3172-83.
155. Kato M, Paranjape T, Muller RU, Nallur S, Gillespie E, Keane K, Esquela-Kerscher A, Weidhaas JB, Slack FJ. The mir-34 microRNA is required for the DNA damage response in vivo in C. elegans and in vitro in human breast cancer cells. Oncogene 2009;28:2419-24.
156. Chaudhry MA, Sachdeva H, Omaruddin RA. Radiation-induced micro-RNA modulation in glioblastoma cells differing in DNA-repair pathways. DNA Cell Biol 2010;29:553-61.
157. He L, He X, Lim LP, de Stanchina E, Xuan Z, Liang Y, Xue W, Zender L, Magnus J, Ridzon D, Jackson AL, Linsley PS, Chen C, Lowe SW, Cleary MA, Hannon GJ. A microRNA component of the p53 tumour suppressor network. Nature 2007;447:1130-4.
158. Taganov KD, Boldin MP, Chang KJ, Baltimore D. NF-kappaB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc Natl Acad Sci U S A 2006;103:12481-6.
159. Zhou R, Hu G, Liu J, Gong AY, Drescher KM, Chen XM. NF-kappaB p65-dependent transactivation of miRNA genes following Cryptosporidium parvum infection stimulates epithelial cell immune responses. PLoS Pathog 2009;5:e1000681.
160. Polytarchou C, Iliopoulos D, Hatziapostolou M, Kottakis F, Maroulakou I, Struhl K, Tsichlis PN. Akt2 regulates all Akt isoforms and promotes resistance to hypoxia through induction of miR-21 upon oxygen deprivation. Cancer Res 2011;71:4720-31.
161. Ruan Q, Wang T, Kameswaran V, Wei Q, Johnson DS, Matschinsky F, Shi W, Chen YH. The microRNA-21-PDCD4 axis prevents type 1 diabetes by blocking pancreatic beta cell death. Proc Natl Acad Sci U S A 2011;108:12030-5.
162. Niu J, Shi Y, Tan G, Yang CH, Fan M, Pfeffer LM, Wu ZH. DNA damage induces NF-κB-dependent microRNA-21 up-regulation and promotes breast cancer cell invasion. J Biol Chem 2012;287:21783-95.
163. Niu J, Xue A, Chi Y, Xue J, Wang W, Zhao Z, Fan M, Yang CH, Shao ZM, Pfeffer LM, Wu J, Wu ZH. Induction of miRNA-181a by genotoxic treatments promotes chemotherapeutic resistance and metastasis in breast cancer. Oncogene 2016;35:1302-13.
164. Farmer H, McCabe N, Lord CJ, Tutt ANJ, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C, Martin NMB, Jackson SP, Smith GC, Ashworth A. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005;434:917-21.
165. Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ, Helleday T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005;434:913-7.
166. Anders CK, Winer EP, Ford JM, Dent R, Silver DP, Sledge GW, Carey LA. Poly(ADP-Ribose) polymerase inhibition: "targeted" therapy for triple-negative breast cancer. Clin Cancer Res 2010;16:4702-10.
167. Plummer ER. Inhibition of poly(ADP-ribose) polymerase in cancer. Curr Opin Pharmacol 2006;6:364-8.
168. Stilmann M, Hinz M, Arslan SC, Zimmer A, Schreiber V, Scheidereit C. A nuclear poly(ADP-ribose)-dependent signalosome confers DNA damage-induced IkappaB kinase activation. Mol Cell 2009;36:365-78.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Wang W, Mani AM, Wu ZH. DNA damage-induced nuclear factor-kappa B activation and its roles in cancer progression. J Cancer Metastasis Treat 2017;3:45-59. http://dx.doi.org/10.20517/2394-4722.2017.03
AMA Style
Wang W, Mani AM, Wu ZH. DNA damage-induced nuclear factor-kappa B activation and its roles in cancer progression. Journal of Cancer Metastasis and Treatment. 2017; 3: 45-59. http://dx.doi.org/10.20517/2394-4722.2017.03
Chicago/Turabian Style
Wang, Wei, Arul M. Mani, Zhao-Hui Wu. 2017. "DNA damage-induced nuclear factor-kappa B activation and its roles in cancer progression" Journal of Cancer Metastasis and Treatment. 3: 45-59. http://dx.doi.org/10.20517/2394-4722.2017.03
ACS Style
Wang, W.; Mani AM.; Wu Z.H. DNA damage-induced nuclear factor-kappa B activation and its roles in cancer progression. J. Cancer. Metastasis. Treat. 2017, 3, 45-59. http://dx.doi.org/10.20517/2394-4722.2017.03
About This Article
Copyright

Author Biographies

Data & Comments
Data


 Cite This Article 13 clicks
Cite This Article 13 clicks

 Like This Article 23
likes
Like This Article 23
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.