
Muscle mitochondria and oxidative metabolism as targets against cancer cachexia


Abstract
Cancer cachexia is a debilitating syndrome mainly characterized by muscle and fat wasting, leading to the progressive loss of body weight and complicating the management of cancer patient. In particular, the loss of muscle weight is a negative prognostic factor, being associated with chemotherapy toxicity and reduced survival. Increased inflammation and protein dysmetabolism are some of the impairments that lead to muscle wasting in cancer patients. Together with these alterations, tumor growth and chemotherapy administration may affect mitochondrial function, impinging on the muscle energy metabolism. Indeed, therapeutic approaches poised to correct both hypercatabolism and mitochondrial alterations could be effective in preventing cancer-induced muscle wasting. Among the non-pharmacological approaches, exercise training is one of the best modulator of muscle physiology able to impinge on both protein and energy metabolism. However, the wasting phenotype that characterizes cancer patients could be not compatible with physical training, prompting the development of different strategies to improve muscle metabolism. The aim of this mini-review is to discuss both the beneficial effects and the limitations of exercise training in cancer cachexia and the adoption of drugs able to modulate exercise-induced pathways.
Keywords
Introduction
Cachexia is a metabolic disorder that occurs in different chronic diseases, including cancer, heart failure, kidney disease, chronic obstructive pulmonary disease, sepsis and rheumatoid arthritis[1]. The prevalence of cachexia is particularly high in cancer patients, ranging from 50% to 80%, depending on tumor type[2]. Cancer cachexia is characterized by body weight loss, systemic inflammation and metabolic alterations. The occurrence of this syndrome affects patient’s quality of life and increases both morbidity and mortality due to cardiovascular complications, immune disorders and nutritional deficiency[3,4]. Body weight loss is mainly due to the consistent depletion of muscle and fat mass, that is frequently worsened by chemotherapy toxicity[5]. Humoral mediators and altered energy balance activate proteolysis in the skeletal muscle of cancer hosts, ultimately leading to protein and organelle disposal[6]. However, the specific blockade of intracellular muscle proteolytic systems does not prove effective in counteracting cancer-induced wasting[7-9], focusing the investigation to upstream pathways that regulate muscle metabolism and energetics. In particular, several lines of evidence show that alterations of the energy metabolism and mitochondrial impairments may contribute to the onset and progression of muscle wasting in cancer[10]. In this regard, cancer hosts are characterized by low nutrient intake and increased energy expenditure[1]. Whereas the former is induced by anorexia, dysphagia and/or altered absorption, due to chemotherapy or to tumors with esophageal/gastrointestinal localization[11], the latter is enhanced by tumor metabolism, inflammation and mitochondrial alterations[1]. Notably, one of the first evidences of energy wasting in cancer cachexia derive from studies on uncoupling proteins (UCPs), whose expression is increased in the skeletal muscle and adipose tissue of tumor-bearing animals and cancer patients[12-15]. Along this line, a time-course analysis in lewis lung carcinoma (LLC)-bearing mice has shown that mitochondrial ROS emission, mitochondrial degradation and respiratory function are impaired prior to the loss of muscle mass in the host mice and, interestingly, some of these alterations appear early after tumor implantation[16]. A proteomic profiling of both skeletal and cardiac muscles of C26-bearing mice, another syngeneic model of cancer cachexia, has revealed an impaired expression of proteins involved in energy homeostasis and mitochondrial function[17]. Consistently, cachectic cancer patients show altered levels of proteins accepted as markers of mitophagy, such as Parkin, PTEN-induced putative kinase 1 (PINK1) and BCL2/adenovirus E1B 19 kDa protein-interacting protein 3 (BNIP3), and mitochondrial dynamics, such as mitofusin 2 (Mfn2) and mitochondrial fission 1 protein (Fis1)[18,19]. In addition, it is well established that anti-cancer treatments alter mitochondrial homeostasis. In this regard, healthy mice treated with FOLFOX or FOLFIRI (the combination of 5-fluorouracil and leucovorin with either oxaliplatin or irinotecan, respectively) present with reduced mitochondrial mass and oxidative capacity in the skeletal muscle[20]. Similar results have been reported in C26-bearing mice treated with chemotherapy (oxaliplatin and 5-fluorouracil)[21], showing that anti-cancer treatment significantly increased the lifespan of C26-bearing mice but exacerbate muscle wasting as compared to untreated C26 hosts[21]. Such wasting effect has been associated with reduced peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α) and cytochrome c expression and with increased markers of mitophagy, together with impaired oxidative capacity and ATP levels[21]. Using a proteomic approach, alterations of the tricarboxylic acid (TCA) cycle and impaired expression of markers of mitochondrial fusion, fission and biogenesis have been reported in both untreated and chemotherapy-treated C26-bearing mice[22]. A metabolic profiling performed in the same C26 hosts exposed to chemotherapy shows that glycolysis and β-oxidation are also impaired[23].
Overall, since muscle metabolic phenotype in cancer hosts is severely impaired by both tumor growth and chemotherapy, strategies targeting mitochondria and/or improving the oxidative capacity could usefully integrate a multimodal therapy for cancer cachexia.
Exercise as a modulator of muscle metabolism
Physical activity is associated with several metabolic adaptations that particularly affect the skeletal muscle[24]. Depending on the type and frequency of exercise, different targets are preferentially regulated. Acute exercise leads to transient muscle molecular changes (e.g., temporary increase in specific mRNA expression), whereas chronic exercise induces physiological adaptations that could modify muscle phenotype[25]. Regarding the differences in terms of exercise type, resistance training imposes a low-repetition and high-intensity demand on the skeletal muscle, whereas endurance exercise does the opposite[26], resulting in specific skeletal muscle adaptations. Thus, resistance training leads to the induction of myofibrillar protein synthesis and increased anaerobic capacity, whereas endurance exercise sessions mainly lead to improved mitochondrial density and function, oxygen delivery and uptake, anti-oxidant defenses and resistance to fatigue[25]. The beneficial effects of exercise have been tested in different chronic conditions, including coronary heart disease, type 2 diabetes mellitus, obesity and aging[24,27]. Notably, both exercise types exert beneficial effects in cachectic animals. Resistance training by ladder climbing has been reported to relieve body weight loss, muscle wasting and inflammation in tumor-bearing rats[28,29] and to increase the expression of factors associated with myogenesis in cachectic mice[30]. In Apc+/Min mice overexpressing IL-6, endurance exercise prevents body weight loss, improves insulin sensitivity, increases mitochondrial biogenesis and muscle oxidative capacity and restores normal signaling through Akt and 5’ AMP-activated protein kinase (AMPK)[31,32]. Endurance exercise by voluntary wheel running is effective also in C26-bearing mice, reducing body weight loss, muscle wasting and the expression of markers of autophagy and proteasome-dependent degradation[33]. However, the combination of the two types of exercise could result in better outcomes in cancer hosts. This was suggested by the results of an exercise protocol in which the slope was increased in a mild intensity run protocol using a motorized wheel. Indeed, this protocol mixed endurance exercise, due to the moderate and continuous training, to the resistance exercise, resulting by the increased hill as compared to the flat treadmill run. Notably, C26 hosts performing this exercise protocol have shown improved muscle wasting and strength, together with reduced autophagy and oxidative stress markers[34]. On the contrary, endurance exercise with treadmill in a flat mode does not succeed in improving cachexia in C26 hosts[35]. The effect of exercise trough a motorized wheel has also been tested in a model of cancer (C26 tumor) and chemotherapy-induced muscle wasting, partially preventing the loss of muscle mass and strength[21]. These improvements are associated with a partial restoration of mitochondrial homeostasis[21]. Notably, exercise increases mitochondrial mass, normalizes the levels of PINK1, BNIP3 and Mfn2, improves the succinate dehydrogenase (SDH) activity and increases energy stores[21]. In a recent study, the resistance exercise component has been further increased by combining the motorized wheel to the ladder climbing. This combined protocol relieves muscle wasting in C26-bearing mice, reducing autophagy markers and increasing muscle oxidative metabolism[36].
The effects of physical training in cancer patients may vary depending on tumor type, stage and exercise modalities. A systematic review analyzing the effectiveness of aerobic exercise during chemotherapy treatment in cancer patients, has shown beneficial effects in terms of both quality of life and muscle function[37]. In chemotherapy-treated breast cancer patients, exercise increases muscle fiber cross-sectional area, citrate synthase activity, mitochondrial complex protein levels and muscle capillarization[38]. Supervised progressive resistance training increases muscle strength in pancreatic cancer hosts[39]; similarly, it decreases fatigue and improves quality of life in head and neck cancer patients[40]. Although these encouraging data, exercise has some limitations in terms of feasibility and patient compliance. In this regard, a study in a cohort of 196 cancer patients with established cachexia has shown a scarce adherence to exercise programs, with low ability to perform either aerobic or resistance training[41]. Along this line, cancer patients with severe or refractory cachexia have a lack of interest in, or cannot cope with, exercise or do not complete the initially planned program[42]. In addition, exercise is not free of risks and may harm cancer patients presenting with co-morbities (anemia or cardiac dysfunctions)[43] or with excessive loss of muscle mass and low energy availability. Depending on exercise type and duration, physical activity may worsen body and muscle wasting also in cachectic animals. Notably, in C26-bearing mice 2-week-long treadmill run exacerbates body and muscle wasting as compared to sedentary tumor bearers[35]. In addition, exercise performed in late-stage cachexia reduces the survival of C26-bearing mice exposed to chemotherapy[21]. Given these premises, strategies that circumvent the limitations of exercise or improve exercise capacity could be useful for cancer hosts who are intolerant to physical training[44][Figure 1].
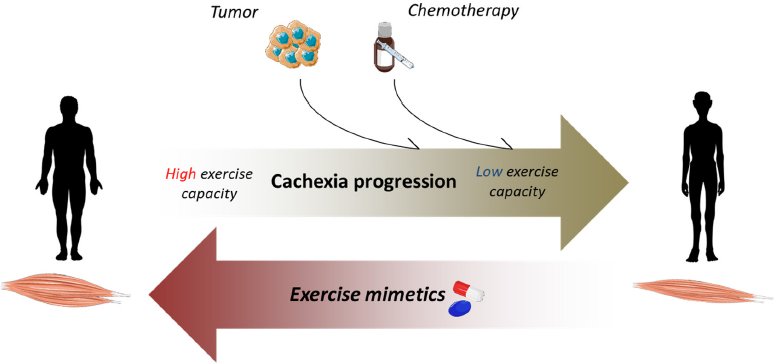
Figure 1. Exercise mimetics may positively affect cachexia progression and low exercise performance. In cancer patients, tumor growth and chemotherapy may induce anemia, cardiac dysfunction and mitochondrial alterations, leading to loss of muscle mass and reduced exercise performance. Exercise-like drugs may relieve muscle wasting and/or support cancer patients practicing exercise
Targeting exercise-induced pathways and muscle metabolism as therapeutic strategy
One of the most relevant player in the exercise-induced adaptations in the skeletal muscle is the co-transcription factor PGC-1α, that responds to energy demand by enhancing the transcription of genes involved in mitochondrial biogenesis and turnover, leading to increased muscle oxidative capacity[44,45]. In this regard, the ablation of PGC-1α in mice has been shown to blunt the increase of autophagy and mitophagy induced by exercise, to decrease mitochondrial mass and to impair exercise performance[45,46]. Conversely, PGC-1α overexpression increases mitochondrial content, fast-to-slow muscle fiber switch and induces a set of genes associated with energy metabolism[47,48]. Moreover, PGC-1α transgenic animals show an improvement of satellite cell activation and proliferative potential[49]. Indeed, PGC-1α plays an important role in modulating muscle homeostasis and its downregulation is correlated with different wasting conditions, such as aging, muscle disuse and denervation[50-52]. In aged mice, PGC-1α overexpression has been shown to counteract the loss of muscle mitochondrial enzymes, to increase markers of autophagy and to preserve both neuromuscular junctions[47] and exercise capacity[53]. In muscle disuse and denervation, increased PGC-1α expression prevents muscle atrophy and the activation of catabolic pathways[50,52]. In cancer cachexia, muscle PGC-1α protein expression is reduced in different animal models and such impairment does not always reflect a reduction of the transcript level[54]. Otherwise, some studies also reports an increase of PGC-1α gene expression[54]. Independently from PGC-1α, muscle mitochondrial mass and metabolism are severely affected in tumor-bearing animals[10], suggesting that PGC-1α is not the only determining factor in impaired mitochondrial function. In cachectic animals, conflicting results are also reported regarding PGC-1α overexpression. Whereas Wang et al.[55] demonstrate that the muscle-specific upregulation of PGC-1α does not protect LLC-bearing mice from muscle atrophy in the same animal model Pin et al.[35] show increased muscle mass and reduced atrogin-1 and muscle RING-finger protein-1 (MuRF-1) gene expression as compared to wild-type tumor bearers. However, both the studies suggest that forcing the expression of PGC-1α in the skeletal muscle could have some limitations, such as the increase in tumor mass[35,55].
The effects of boosting mitochondrial biogenesis have been tested also in cancer and chemotherapy-induced muscle wasting. In this condition, PGC-1α overexpression does not improve muscle mass, whereas this effect is achieved by moderate exercise[21]. This could suggest that enhancing mitochondrial biogenesis is not sufficient to mimic the effects of exercise and that it does not necessarily reflect an increase of mitochondrial function, both being affected in chemotherapy-treated tumor hosts[21].
The modulations triggered by exercise may be mimicked by pharmacological and dietary compounds[44,56]. Some of them have been extensively studied, such as 5-aminoimidazole-4-carboxyamide ribonucleoside (AICAR), glitazones, metformin and sirtuin 1 (SIRT1) activators, also reporting their effectiveness in preventing cancer-induced muscle wasting[10,44]. Also erythropoietin (EPO), an endogenous cytokine essential for the growth and differentiation of red blood progenitor cells, has shown some exercise-like effects that go beyond the increase in oxygen delivery. Indeed, the EPO receptor (EpoR) is expressed in different tissues other than hematopoietic cells, such as heart, skeletal muscle, adipose tissue, brain and pancreas[57]. In this regard, EPO stimulates SIRT1 signaling in human cardiomyocytes treated with doxorubicin, increasing the levels of PGC-1α, nuclear respiratory factor 1 (NRF1), citrate synthase and cytochrome c oxidase IV[58]. EPO exposure of C2C12 myocyte cultures and primary skeletal myoblasts in vitro results in increased mitochondrial mass, PGC-1α levels, citrate synthase activity and oxygen consumption rate[59]. Similarly, transgenic mice expressing high levels of EPO show increased proportion of oxidative fibers and improved mitochondrial activity in the skeletal muscle, together with increased PGC-1α and AMPK activation[59]. Recombinant EPO administration to humans also results in increased mitochondrial oxidative phosphorylation and electron transport capacity[60]. Consistently, LLC-bearing mice treated with EPO show increased SDH activity, ATP content and PGC-1α expression in the skeletal muscle[35]. These effects are enhanced by combining exercise training and EPO, protecting the mitochondrial compartment and resulting in increased muscle strength as compared to untreated sedentary cancer hosts[35].
Another modulator of muscle metabolism is trimetazidine (TMZ). The effects of TMZ are primarily related to the ability to block β-oxidation, by inhibiting the 3-ketoacyl-CoA thiolase (3-KAT) activity in the mitochondrial matrix[61]. This inhibition results in a metabolic shift towards glucose oxidation, optimizing oxygen utilization and decreasing lactate levels[62]. TMZ is used to treat chronic stable angina and ischemic cardiomyopathy[63], reducing symptoms and improving exercise performance[63,64]. The effects on patients with cardiovascular diseases could be also related to the protection of the mitochondrial compartment. In this regard, TMZ is able to correct the toxicity induced by palmitate in cultured cardiomyocytes, increasing mitochondria mass, volume and function[65]. Positive effects have been achieved by TMZ also on C2C12 myotubes exposed to TNF-α or to growth factor deprivation. Notably, TMZ induces the activation of anabolic pathways and reduces the expression of atrogin-1 and MuRF1, protecting C2C12 myotubes from atrophy[66]. Of relevance, TMZ is also able to induce autophagy in vitro[66]. Moving to in vivo models, TMZ improves muscle strength of aged mice, coupled with a muscle metabolic shift towards oxidative metabolism, suggested by increased levels of the slow myosin heavy chain (MyHC) isoform[67]. Similarly to aged muscle, TMZ induces exercise-like effects also in the skeletal muscle of tumor-bearing animals. In this regard, TMZ administration partially protects C26-bearing mice from muscle atrophy (myofiber cross sectional area) and dysfunction, increasing slow MyHC content, SDH-positive myofibers and markers associated with mitochondrial mass and biogenesis[68]. An increase in vascular endothelial cadherin (Ve-cadherin) and vascular endothelial growth factor (VEGF) expression, two known markers of angiogenesis, has also been detected in tumor-bearing mice after TMZ administration[68]. Of relevance, TMZ seems to induce its effects quickly in the skeletal muscle. Indeed, 15 min ex-vivo TMZ treatment of EDL muscle (mostly composed by glycolytic fibers) is enough to reduce both contraction and relaxation rates[68], favoring the occurrence of a phenotype typical of slow-twitch muscle[69].
Exercise is also able to activate myogenesis, impinging on satellite cells (SCs) proliferation and differentiation[70]. Accordingly, TMZ treatment modulates the behavior of differentiating murine myoblast, increasing fusion capacity, markers of myogenesis and MyHC levels[71]. These effects are associated with increased expression of metabolic regulators such as PGC-1α and activated AMPK, suggesting an increase in oxidative metabolism[71]. Similar results have been obtained in isolated murine SCs, in which TMZ increases some markers of differentiation as compared to control[71]. Myogenesis is impaired in cachectic animals and cancer patients, likely contributing to muscle wasting[72,73]. In this regard, the effect of TMZ on myogenesis markers has been tested on C26-bearing mice, showing increased levels of MyoD and Myogenin[71]. Such results suggest that TMZ could potentially protect against myogenesis impairments.
Conclusion
The research on the etiopathogenesis of cancer cachexia has unraveled different metabolic alterations that lead to the wasting phenotype of cancer patients. Exercise training is one of the best approaches able to correct the metabolic impairments of tumor hosts, such as inflammation, hypercatabolism and energy deficit. However, being the skeletal muscle severely affected in cancer cachexia, often the physical (and psychological) state of cancer patients is not permissive for practicing exercise. Also due to these reasons the search for pharmacological strategies able to modulate exercise-induced pathways is rapidly growing. In this regard, exercise beneficial effects in cancer cachexia could depend, partially at least, on the expression of PGC-1α. Consistently, drugs able to enhance the expression of this co-transcription factor, together with strategies that increase mitochondrial function, could be effective in improving muscle wasting [Figure 2]. However, beyond the alterations of the mitochondrial homeostasis, muscle depletion relies also on inflammation, activation of catabolic pathways and, frequently, depression of protein synthesis. Along this line, a combinatorial therapy targeting each of these alterations could be the best choice to counteract cancer-induced muscle wasting.
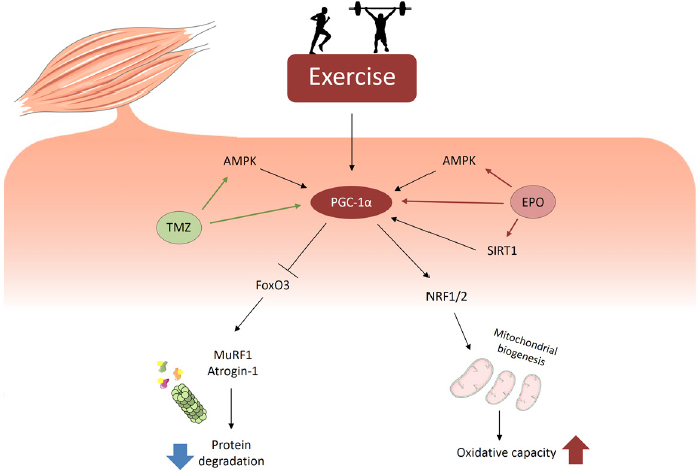
Figure 2. Exercise-like responses in the skeletal muscle may be induced by drugs acting on PGC-1α expression and activity. Exercise modulates different pathways involved in the regulation of protein homeostasis and energy metabolism. During exercise, PGC-1α activation increased mitochondrial biogenesis, favoring oxidative capacity, and inhibits FoxO3, reducing proteasome-dependent degradation. Similarly to exercise, EPO and TMZ are able to activate energy sensors, such as AMPK and SIRT1, that induce PGC-1α and its downstream targets
Declarations
Authors’ contributionsWrote the manuscript: Ballarò R
Discussed and revised the manuscript: Penna F, Ferraro E, Costelli P
Availability of data and materialsNot applicable.
Financial support and sponsorshipNone.
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2019.
REFERENCES
1. Baracos VE, Martin L, Korc M, Guttridge DC, Fearon KCH. Cancer-associated cachexia. Nat Rev Dis Prim 2018;4:1-18.
2. von Haehling S, Anker MS, Anker SD. Prevalence and clinical impact of cachexia in chronic illness in Europe, USA, and Japan: facts and numbers update 2016. J Cachexia Sarcopenia Muscle 2016;7:507-9.
3. Kalantar-Zadeh K, Rhee C, Sim JJ, Stenvinkel P, Anker SD, et al. Why cachexia kills: Examining the causality of poor outcomes in wasting conditions. J Cachexia Sarcopenia Muscle 2013;4:89-94.
4. Evans WJ, Morley JE, Argilés J, Bales C, Baracos V, et al. Cachexia:a new definition. Clin Nutr 2008;27:793-9.
5. Pin F, Couch ME, Bonetto A. Preservation of muscle mass as a strategy to reduce the toxic effects of cancer chemotherapy on body composition. Curr Opin Support Palliat Care 2018;12:420-6.
6. Argilés JM, Busquets S, Stemmler B, López-Soriano FJ. Cancer cachexia: understanding the molecular basis. Nat Rev Cancer 2014;14:754-62.
7. Penna F, Ballarò R, Martinez-cristobal P, Sebastian D, Busquets S, et al. Autophagy exacerbates muscle wasting in cancer cachexia and impairs mitochondrial function. J Mol Biol 2019:2674-86.
8. Penna F, Costamagna D, Pin F, Camperi A, Fanzani A, et al. Autophagic degradation contributes to muscle wasting in cancer cachexia. Am J Pathol 2013;182:1367-78.
9. Penna F, Bonetto A, Aversa Z, Minero VG, Rossi Fanelli F, et al. Effect of the specific proteasome inhibitor bortezomib on cancer-related muscle wasting. J Cachexia Sarcopenia Muscle 2015;7:345-54.
10. Penna F, Ballarò R, Beltrá M, De Lucia S, Costelli P. Modulating metabolism to improve cancer-induced muscle wasting. Oxid Med Cell Longev 2018;2018:7153610.
11. von Meyenfeldt M. Cancer-associated malnutrition: an introduction. Eur J Oncol Nurs 2005;9:35-8.
12. Collins P, Bing C, McCulloch P, Williams G. Muscle UCP-3 mRNA levels are elevated in weight loss associated with gastrointestinal adenocarcinoma in humans. Br J Cancer 2002;86:372-5.
13. Julienne CM, Dumas JF, Goupille C, Pinault M, Berri C, et al. Cancer cachexia is associated with a decrease in skeletal muscle mitochondrial oxidative capacities without alteration of ATP production efficiency. J Cachexia Sarcopenia Muscle 2012;3:265-75.
14. Busquets S, Almendro V, Barreiro E, Figueras M, Argilés JM, et al. Activation of UCPs gene expression in skeletal muscle can be independent on both circulating fatty acids and food intake: Involvement of ROS in a model of mouse cancer cachexia. FEBS Lett 2005;579:717-22.
15. Argilés JM, López-Soriano FJ, Busquets S. Muscle wasting in cancer: the role of mitochondria. Curr Opin Clin Nutr Metab Care 2015;18:221-5.
16. Brown JL, Rosa-Caldwell ME, Lee DE, Blackwell TA, Brown LA, et al. Mitochondrial degeneration precedes the development of muscle atrophy in progression of cancer cachexia in tumour bearing mice. J Cachexia Sarcopenia Muscle 2017;8:926-38.
17. Shum AMY, Poljak A, Bentley NL, Turner N, Tan TC, et al. Proteomic profiling of skeletal and cardiac muscle in cancer cachexia: alterations in sarcomeric and mitochondrial protein expression. Oncotarget 2018;9:22001-22.
18. Marzetti E, Lorenzi M, Landi F, Picca A, Rosa F, et al. Altered mitochondrial quality control signaling in muscle of old gastric cancer patients with cachexia. Exp Gerontol 2017;87:92-9.
19. Aversa Z, Pin F, Lucia S, Penna F, Verzaro R, et al. Autophagy is induced in the skeletal muscle of cachectic cancer patients. Sci Rep 2016;6:30340.
20. Barreto R, Waning DL, Gao H, Liu Y, Zimmers TA, et al. Chemotherapy-related cachexia is associated with mitochondrial depletion and the activation of ERK1/2 and p38 MAPKs. Oncotarget 2016;7:43442-60.
21. Ballaro R, Beltra M, De Lucia S, Pin F, Ranjbar K, et al. Moderate exercise in mice improves cancer plus chemotherapy-induced muscle wasting and mitochondrial alterations. FASEB J 2019;33:5482-94.
22. Barreto R, Mandili G, Witzmann FA, Novelli F, Zimmers TA, et al. Cancer and chemotherapy contribute to muscle loss by activating common signaling pathways. Front Physiol 2016;7:1-13.
23. Pin F, Bonetto A, Oconnell T. Cachexia induced by cancer and chemotherapy yield distinct perturbations to energy metabolism. J Cachexia Sarcopenia Muscle 2019;10:140-54.
24. Viña J, Sanchis-Gomar F, Martinez-Bello V, Gomez-Cabrera MC. Exercise acts as a drug; The pharmacological benefits of exercise. Br J Pharmacol 2012;167:1-12.
25. Egan B, Zierath JR. Exercise metabolism and the molecular regulation of skeletal muscle adaptation. Cell Metab 2013;17:162-84.
26. Hardee JP, Counts BR, Carson JA. Understanding the role of exercise in cancer cachexia therapy. Am J Lifestyle Med 2017;13:46-60.
27. Coletti D. Exercise against tumor- and chemotherapy-induced muscle wasting. Biol Eng Med 2018;3:1-5.
28. Padilha CS, Borges FH, Costa Mendes da Silva LE, Frajacomo FTT, Jordao AA, et al. Resistance exercise attenuates skeletal muscle oxidative stress, systemic pro-inflammatory state, and cachexia in Walker-256 tumor-bearing rats. Appl Physiol Nutr Metab 2017;42:916-23.
29. Donatto FF, Neves RX, Rosa FO, Camargo RG, Ribeiro H, et al. Resistance exercise modulates lipid plasma profile and cytokine content in the adipose tissue of tumour-bearing rats. Cytokine 2013;61:426-32.
30. Khamoui AV, Park BS, Kim DH, Yeh MC, Oh SL, et al. Aerobic and resistance training dependent skeletal muscle plasticity in the colon-26 murine model of cancer cachexia. Metabolism 2016;65:685-98.
31. Puppa MJ, White JP, Velázquez KT, Baltgalvis KA, Sato S, et al. The effect of exercise on IL-6-induced cachexia in the ApcMin/+ mouse. J Cachexia Sarcopenia Muscle 2012;3:117-37.
32. White JP, Puppa MJ, Sato S, Gao S, Price RL, et al. IL-6 regulation on skeletal muscle mitochondrial remodeling during cancer cachexia in the ApcMin/+ mouse. Skelet Muscle 2012;2:14.
33. Pigna E, Berardi E, Aulino P, Rizzuto E, Zampieri S, et al. Aerobic Exercise and Pharmacological Treatments Counteract Cachexia by Modulating Autophagy in Colon Cancer. Sci Rep 2016;6:26691.
34. Ballarò R, Penna F, Pin F, Gómez-Cabrera MC, Viña J, et al. Moderate Exercise Improves Experimental Cancer Cachexia by Modulating the Redox Homeostasis. Cancers (Basel) 2019;11:E285.
35. Pin F, Busquets S, Toledo M, Camperi A, Lopez-Soriano FJ, et al. Combination of exercise training and erythropoietin prevents cancer-induced muscle alterations. Oncotarget 2015;6:43202-15.
36. Ranjbar K, Ballaro R, Bover Q, Pin F, Beltra M, et al. Combined exercise training positively affects muscle wasting in tumor-bearing mice. Med Sci Sports Exerc 2019;51:1387-95.
37. Cave J, Paschalis A, Huang CY, West M, Copson E, et al. A systematic review of the safety and efficacy of aerobic exercise during cytotoxic chemotherapy treatment. Support Care Cancer 2018;26:3337-51.
38. Mijwel S, Cardinale DA, Norrbom J, Chapman M, Ivarsson N, et al. Exercise training during chemotherapy preserves skeletal muscle fiber area, capillarization, and mitochondrial content in patients with breast cancer. FASEB J 2018;32:5495-505.
39. Wiskemann J, Clauss D, Tjaden C, Hackert T, Schneider L, et al. Progressive resistance training to impact physical fitness and body weight in pancreatic cancer patients: a randomized controlled trial. Pancreas 2019;48:257-66.
40. Grote M, Maihöfer C, Weigl M, Davies-Knorr P, Belka C. Progressive resistance training in cachectic head and neck cancer patients undergoing radiotherapy: a randomized controlled pilot feasibility trial. Radiat Oncol 2018;13:1-10.
41. Wasley D, Gale N, Roberts S, Backx K, Nelson A, et al. Patients with established cancer cachexia lack the motivation and self-efficacy to undertake regular structured exercise. Psychooncology 2018;27:458-64.
42. Antoun S, Raynard B. Muscle protein anabolism in advanced cancer patients:response to protein and amino acids support, and to physical activity. Ann Oncol 2018;29:ii10-7.
43. Argilés JM, Busquets S, López-Soriano FJ, Costelli P, Penna F. Are there any benefits of exercise training in cancer cachexia? J Cachexia Sarcopenia Muscle 2012;3:73-6.
44. Penna F, Pin F, Ballarò R, Baccino FM, Costelli P. Novel investigational drugs mimicking exercise for the treatment of cachexia. Expert Opin Investig Drugs 2016;25:63-72.
45. Vainshtein A, Tryon LD, Pauly M, Hood DA. Role of PGC-1α during acute exercise-induced autophagy and mitophagy in skeletal muscle. Am J Physiol - Cell Physiol 2015;308:C710-9.
46. Brandt N, Dethlefsen MM, Bangsbo J, Pilegaard H. PGC-1 α and exercise intensity dependent adaptations in mouse skeletal muscle. PLoS One 2017;12:e0185993.
47. Garcia S, Nissanka N, Mareco EA, Rossi S, Peralta S, et al. Overexpression of PGC-1 a in aging muscle enhances a subset of young-like molecular patterns. Aging Cell 2018;17.
48. Lin J, Wu H, Tarr P, Zhang C, Wu Z, et al. Transcriptional co-activator PGC-1 a drives the formation of slow-twitch muscle fibre. Nature 2002;418:797-801.
49. Dinulovic I, Furrer R, Beer M, Ferry A, Cardel B, et al. Muscle PGC-1 α modulates satellite cell number and proliferation by remodeling the stem cell niche. Skelet Muscle 2016;6:39.
50. Cannavino J, Brocca L, Sandri M, Bottinelli R, Pellegrino MA. PGC1- α over-expression prevents metabolic alterations and soleus muscle atrophy in hindlimb unloaded mice. J Physiol 2014;20:4575-89.
52. Sandri M, Lin J, Handschin C, Yang W, Arany ZP, et al. PGC-1alpha protects skeletal muscle from atrophy by suppressing FoxO3 action and atrophy-specific gene transcription. Proc Natl Acad Sci U S A 2006;103:16260-5.
53. Gill JF, Santos G, Schnyder S, Handschin C. PGC-1α affects aging-related changes in muscle and motor function by modulating specific exercise-mediated changes in old mice. Aging Cell 2018;17:1-13.
54. van der Ende M, Grefte S, Plas R, Meijerink J, Witkamp RF, et al. Mitochondrial dynamics in cancer-induced cachexia. Biochim Biophys Acta - Rev Cancer 2018;1870:137-50.
55. Wang X, Pickrell AM, Zimmers TA, Moraes CT. Increase in muscle mitochondrial biogenesis does not prevent muscle loss but increased tumor size in a mouse model of acute cancer-induced cachexia. PLoS One 2012;7:2-7.
56. Vaughan RA, Mermier CM, Bisoffi M, Trujillo KA, Conn CA. Dietary stimulators of the PGC-1 superfamily and mitochondrial biosynthesis in skeletal muscle. A mini-review. J Physiol Biochem 2014;70:271-84.
57. Wang L, Di L, Noguchi CT. Erythropoietin, a novel versatile player regulating energy metabolism beyond the erythroid system. Int J Biol Sci 2014;10:921-39.
58. Cui L, Guo J, Zhang Q, Yin J, Li J, et al. Erythropoietin activates SIRT1 to protect human cardiomyocytes against doxorubicin-induced mitochondrial dysfunction and toxicity. Toxicol Lett 2017;275:28-38.
59. Wang L, Jia Y, Rogers H, Suzuki N, Gassmann M, et al. Erythropoietin contributes to slow oxidative muscle fiber specification via PGC-1α and AMPK activation. Int J Biochem Cell Biol 2014;45:1155-64.
60. Plenge U, Belhage B, Guadalupe-Grau A, Andersen PR, Lundby C, et al. Erythropoietin treatment enhances muscle mitochondria capacity in humans. Front Physiol 2012;3:1-5.
61. Lopaschuk DG, Barr R, Panakkezhum DT, Dyck RJ. Beneficial effects of trimetazidine in ex vivo working ischemic hearts are due to a stimulation of glucose oxidation secondary to inhibition of long-chain 3-ketoacyl coenzyme a thiolase. Circ Res 2003;93:e33-7.
62. Dyck JRB. Malonyl coenzyme a decarboxylase inhibition protects the ischemic heart by inhibiting fatty acid oxidation and stimulating glucose oxidation. Circ Res 2004;94:e78-84.
63. Guarini G, Huqi A, Morrone D, Francesca P, Capozza G, et al. Pharmacotherapy Trimetazidine and Other Metabolic Modifiers. Eur Cardiol 2018;13:104-11.
64. Vitale C, Marazzi G, Pelliccia F, Volterrani M, Cerquetani E, et al. Trimetazidine improves exercise performance in patients with peripheral arterial disease. Pharmacol Res 2011;63:278-83.
65. Kuzmicic J, Parra V, Verdejo HE, López-Crisosto C, Chiong M, et al. Trimetazidine prevents palmitate-induced mitochondrial fission and dysfunction in cultured cardiomyocytes. Biochem Pharmacol 2014;91:323-36.
66. Ferraro E, Giammarioli AM, Caldarola S, Lista P, Feraco A, et al. The metabolic modulator trimetazidine triggers autophagy and counteracts stress-induced atrophy in skeletal muscle myotubes. FEBS J 2013;280:5094-108.
67. Ferraro E, Pin F, Gorini S, Pontecorvo L, Ferri A, et al. Improvement of skeletal muscle performance in ageing by the metabolic modulator Trimetazidine. J Cachexia Sarcopenia Muscle 2016;7:449-57.
68. Molinari F, Pin F, Gorini S, Chiandotto S, Pontecorvo L, et al. The mitochondrial metabolic reprogramming agent trimetazidine as an ‘exercise mimetic’ in cachectic C26-bearing mice. J Cachexia Sarcopenia Muscle 2017;8:954-73.
69. Blaauw B, Schiaffino S, Reggiani C. Mechanisms modulating skeletal muscle phenotype. Compr Physiol 2013;3:1645-87.
70. Bazgir B, Fathi R, Valojerdi MR, Mozdziak P, Asgari A. Satellite cells contribution to exercise mediated muscle hypertrophy and repair. Cell J 2016;18:473-84.
71. Gatta L, Vitiello L, Gorini S, Chiandotto S, Costelli P, et al. Modulating the metabolism by trimetazidine enhances myoblast differentiation and promotes myogenesis in cachectic tumor-bearing c26 mice. Oncotarget 2017;8:113938-56.
72. He WA, Berardi E, Cardillo VM, Acharyya S, Aulino P, et al. NF-κB-mediated Pax7 dysregulation in the muscle microenvironment promotes cancer cachexia. J Clin Invest 2013;123:4821-35.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Ballarò R, Penna F, Ferraro E, Costelli P. Muscle mitochondria and oxidative metabolism as targets against cancer cachexia. J Cancer Metastasis Treat 2019;5:61. http://dx.doi.org/10.20517/2394-4722.2019.003
AMA Style
Ballarò R, Penna F, Ferraro E, Costelli P. Muscle mitochondria and oxidative metabolism as targets against cancer cachexia. Journal of Cancer Metastasis and Treatment. 2019; 5: 61. http://dx.doi.org/10.20517/2394-4722.2019.003
Chicago/Turabian Style
Ballarò, Riccardo, Fabio Penna, Elisabetta Ferraro, Paola Costelli. 2019. "Muscle mitochondria and oxidative metabolism as targets against cancer cachexia" Journal of Cancer Metastasis and Treatment. 5: 61. http://dx.doi.org/10.20517/2394-4722.2019.003
ACS Style
Ballarò, R.; Penna F.; Ferraro E.; Costelli P. Muscle mitochondria and oxidative metabolism as targets against cancer cachexia. J. Cancer. Metastasis. Treat. 2019, 5, 61. http://dx.doi.org/10.20517/2394-4722.2019.003
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 16 clicks
Cite This Article 16 clicks

 Like This Article 0
likes
Like This Article 0
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.