
Phytochemicals and cancer chemoprevention

Abstract
The unending morbidity and mortality that results from cancer, as well as adverse reactions due to chemotherapy and the enormous economic burden of treatment and hospitalization, advocates for the necessity of chemopreventive measures. Cancer chemoprevention refers to the use of agents capable of reversing, reducing, or slowing down the pathology of cancer at various stages. Fortunately, a few therapeutic drugs with relatively low toxicity (e.g., tamoxifen, finasteride), and a sparse number of vaccines (hepatitis B, HPV), are used to prevent specific cancers. In the general population, however, therapeutic options for cancer prevention are not common. Nonetheless, it is generally agreed that diet affects the genesis of cancer, and phytochemicals have the capacity of functioning as cancer chemoprevention agents. This is supported by epidemiological studies and clearly documented with animal models designed to mimic human carcinogenesis. Additionally, some public health strategies, such as recommendations for greater consumption of fruits and vegetables, reflect the merits of cancer chemoprevention. Here, we focus on some well-established natural product cancer chemopreventive agents, including resveratrol (grapes), epigallocatechin-3-gallate (green tea), sulforaphane (cruciferous vegetables), anthocyanins (grapes and berries), curcumin (turmeric), silibinin (milk thistle), and lycopene (tomatoes). As aptly demonstrated by genomic analysis and other methods, the mechanistic underpinning is variable and complex. In addition, responses may be mediated through indirect mechanisms, such as interaction with the microbiome. Furthermore, ancillary applications of chemopreventive agents are worthy of consideration, such as management of sequelae induced by chemotherapy. Recognizing the loss of millions of cancer patients every year, it is obvious that negating malignant metastatic conditions remains of paramount importance. In meeting this objective, cancer chemoprevention offers great promise.
Keywords
Introduction
Overall, cancer is the second leading cause of death in the US and a major public health issue throughout the world. Approximately 1,762,450 new cancer cases are estimated in the US in 2019[1], and 18.1 million new cases worldwide. In 2018, 9.6 million deaths were attributed to this disease[2]. Given these astounding demographics, and the associated pain, suffering and economic burden, the scientific and medical community continuously strive for better treatment options, improved palliative care, and effective preventative strategies. In this context, the role of diet in cancer has attracted considerable attention. This is especially compelling given that epidemiological studies have demonstrated regular consumption of phytochemicals from dietary sources like fruits, vegetables, herbs, and teas is associated with reduced risk of chronic diseases including cancer, cardiovascular disease and inflammatory disorders[3,4].
Cancer chemoprevention can be classified into primary, secondary, and tertiary measures. Populations with no overt cancer risk factors, or those ostensibly at high risk due to factors such as successful surgical resection or family history, can be grouped under primary measures. Patients with pre-malignant lesions bearing risk of progressing to an invasive cancer (e.g., ductal carcinoma in situ) can be grouped under secondary measures. In these cases, standard protocols of chemopreventive practice would be highly desirable, but scarcely come into play[5]. People in primary and secondary chemoprevention categories may be advised or decide on their own to increase dietary phytochemical consumption or to use over the counter products such as non-steroidal anti-inflammatory drugs. Finally, tertiary measures can be considered for patients with cancer relapses[6]. A unique example of a tertiary chemopreventive measure is the administration of tamoxifen (or structural relatives), or aromatase inhibitors, for patients diagnosed with breast cancer[7].
Around two and half thousand years ago, Hippocrates advised “let food be thy medicine and medicine be thy food”. This remains a powerful statement, as graphically illustrated in Figure 1. An inverse relationship between adequate fruit and vegetable consumption and cancer incidence has been established. In fact, it has been suggested that cancer incidence could be reduced by over 50% if people consume at least five servings of fruits and vegetables per day[9]. On the other hand, obesity is generally associated with poor health and chronic illness, and there are certain foods that can act as carcinogens and initiate tumor formation.

Figure 1. Phytochemicals and their chemical structures: anthocyanins in grapes, ellagic acid in raspberry, genistein in soy, 3,3’-diindoylmethane in broccoli, curcumin in turmeric, EGCG in tea, silibinin in milk thistle, and artemisinin in sweet wormwood. Reprinted/adapted by permission from Springer Nature Customer Service Centre GmbH: Springer Nature, Natural Products for Cancer Chemoprevention. Single Compounds and Combinations, by Pezzuto and Vang[8] ©2020
Secondary metabolites (phytochemicals) are typically generated in plants to afford protection against external threats such as UV, fungal infection, and the generation of free radicals. The compounds so produced show a remarkable array of structural diversity. Notably, ingestion of these phytochemicals provide human beings with protective effects as well[10], perhaps by reducing oxidative stress (ROS) and inflammation[3,11]. However, the mechanisms by which phytochemicals function in a chemopreventive capacity are certainly intricate and multifaceted, as described to some extent in this review.
Interestingly, cancer and aging share several hallmarks in terms of the genetic pathways and biochemical processes. For example, DNA repair mechanisms are affected by ROS and this may result in the deregulation of signaling pathways such as p53 and nuclear factor-κB (NF-κB). In turn, such deregulation may accelerate aging and cancer development[12]. In principal, antioxidants sourced from phytochemicals may neutralize ROS and attenuate oxidative stress[13]. Further, as deregulation of signaling pathways is involved in progression of inflammatory diseases, modulation of these processes by phytochemicals may down-regulate proinflammatory factors[14]. Considering the general safety of dietary phytochemicals, especially compared with narrow therapeutic index chemotherapeutic agents, the potential merit of chemoprevention is obvious.
This review encompasses studies involving the dietary role of phytochemicals including curcumin (turmeric), epigallocatechin gallate (green tea), resveratrol (grapes), anthocyanidin (grapes and berries), sulforaphane (cruciferous vegetables), silibinin (herb milk thistle), and lycopene (tomatoes). Mechanisms of alleviating multiple pathological conditions, such as oxidative stress, epigenetic alteration, angiogenesis, chronic inflammation, and effects on stem cell transformation are taken into account. Finally, some thoughts are provided in regard to future directions.
Resveratrol
Chemical properties of resveratrol
Resveratrol (3,5,4ʹ-trihydroxy-trans-stilbene, MW: 228.25 g/mol) is a naturally occurring stilbene with two phenolic rings connected by an ethylene group[15]. It is a phytoalexin mainly synthesized as a protective mechanism in plants in response to environmental stress including fungal infection, UV radiation, and chemical exposure[16]. The dominant dietary source is grapes and grape products[17], but more recently, approximately 70 species of plants have been reported to produce resveratrol[18]. As such, in an average diet, relatively small amounts of resveratrol can be found in peanuts (Arachis hypogaea), blackberries (Morus spp.) and blueberries (Vaccinium spp.)[19,20]. Red wine is a major source of resveratrol in the Mediterranean diet, as grape (Vitis vinifera) is a rich source of resveratrol. Specifically, resveratrol is found in seeds, skin, woody parts, and petioles. Therefore, red wine generally has a higher content of resveratrol than white wine, since, during the production of the wine, parts of grapes in which resveratrol is present are macerated for a longer period of time[21]. During grape fermentation, the formation of alcohol facilitates the solubility of resveratrol which further leads to its extraction.
Polygonum cuspidatum is an extremely rich source of resveratrol and used as a therapeutic regimen for cardiovascular diseases in Chinese and Japanese traditional medicine practice[22]. Similarly, the rhizome of Veratrum formosanum, containing abundant resveratrol, has been applied to treat hypertension in East Asia[23].
Pharmacokinetic properties of resveratrol
Resveratrol is primarily metabolized by phase II enzymes in the liver. Through enterohepatic transport in bile, some of the compound returns to the small intestine[24]. Moreover, resveratrol can stimulate its own metabolism by increasing the action of phase II hepatic detoxifying enzymes[25]. Although trans-piceid, the naturally occurring glucoside, exhibits biological activities, glucuronide metabolites of resveratrol in humans seem to be less active.
The high rate of resveratrol metabolism produces conjugated sulfates and glucuronides which maintain some biological activity[26]. Although metabolites can differ in their nature and quantity between subjects due to inter-individual variability, there are major five types of metabolites in the human urine: two isomeric forms of resveratrol monoglucuronide, resveratrol monosulfate, monoglucuronide dihydroresveratrol and monosulfate dihydroresveratrol[27,28]. In human urine samples, cis metabolites have been found, mainly as cis-resveratrol-4ʹ-sulfate, cis-resveratrol-4ʹ-O-glucuronide and cis-resveratrol-3-O-glucuronide[29,30]. Even though the cis-isomer displayed comparable activities to the trans-isomer in some experimental settings[31,32], the trans-isomer is generally considered dominant and exerts greater activity. Other dietary components may influence metabolism. For example, quercetin has the potential to inhibit resveratrol glucuronidation and sulfation in the duodenal and liver, thus increasing bioavailability[33].
Biological activities of resveratrol
Spearheaded by conceptualization of the “French Paradox”, the potential health benefits of phenolic compounds present in wine and grapes have been extensively studied. As a brief background, in the northern region of France, the dietary intake of saturated fat is relatively high. However, relative to other parts of the world where a similar amount of high saturated fat is consumed, the mortality due to coronary heart disease is reduced. This phenomenon, termed the “French Paradox”, was attributed to the relatively high wine consumption of the French[34]. This possible benefit of wine was not ascribed to alcohol content, since alternative alcoholic beverages such as beer were not perceived to be effective in this regard. In turn, this led to speculation regarding the effectiveness of chemical constituents in wine other than alcohol. It is known that grapes, and consequently wine, contain scores of phytochemicals[35], but a conundrum exists since these components are also found in other sectors of the diet. Alas, when the cancer chemopreventive potential of resveratrol was first described[36], it was recognized this is a compound uniquely associated with the grape (and wine), and, in fact, grapes and wine are the dominant dietary source of this biologically active compound. Thus, resveratrol was perceived by some as a key to the “French Paradox” and many studies followed to explore broader biologic potential[37].
Because of its physical and chemical properties, resveratrol can either interact with receptors present on the cell surface or move passively through cell membranes. Therefore, at the cellular level, action may be initiated by either triggering signaling pathways when binding to the cell membrane receptors, or by facilitating intracellular mechanisms[38]. Accordingly, as discussed in previous reviews[39,40], resveratrol is capable of mediating a myriad of responses. For example, resveratrol is known to down-regulate pro-inflammatory factors [Figure 2], thereby exhibiting an anti-inflammatory response[41]. During the initial stage of an anti-inflammatory response, polymorphonuclear leukocytes play a key role in the process. Resveratrol abates the inflammatory responses initiated by calcium ionophore A23187, fMLP, or component fragment C5a[42]. Inducible nitric oxide synthase (iNOS) activates macrophages, and resveratrol has been shown to decrease the production of iNOS[43,44]. Resveratrol also impedes pro-inflammatory signaling which leads to inhibition of adenosine nucleotide secretion by activated platelets and reduces neutrophil functions through inhibition of P2 and PAP receptor signaling via mitogen-activated protein kinase (MAPK) and c-Jun N-terminal kinase (JNK)[45]. Resveratrol induces extracellular signaling regulated kinases (ERK), p38 MAPK and JNK in the mouse epidermal cells resulting in the phosphorylation of serine 15 of p53, a tumor suppresser gene[46]. Further, resveratrol inhibits macrophage expression of EMMPRIN by inducing PPAR-γ[47] and inhibits NF-κB[48]. In addition, many other studies have confirmed that resveratrol inhibits activation by tumor necrosis factor-α (TNF-α)[49-51].
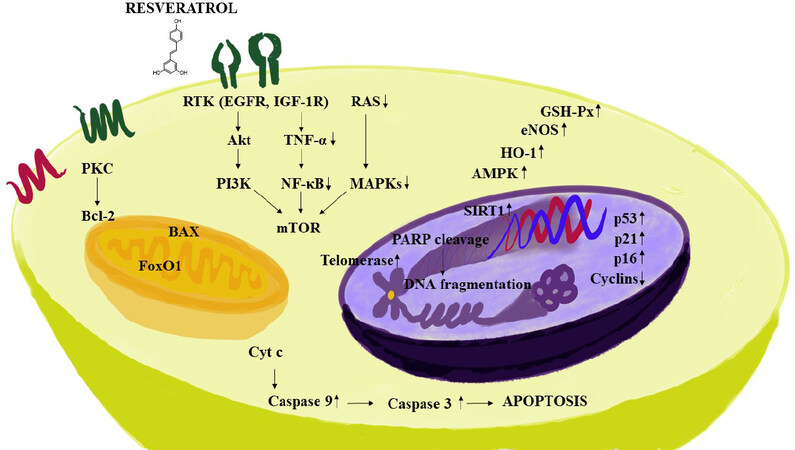
Figure 2. Some mechanisms of action mediated by resveratrol. Resveratrol exhibits an anti-inflammatory response by down-regulating pro-inflammatory factors. The compound decreases survival related proteins including phosphatidylinositol-3-kinase (PI3K) and Akt. Further, signaling cascades are impeded by down-regulating NF-κB, MAPKs and TNF-α, resulting in the inhibition of mTOR. In addition, resveratrol decreases the expression of anti-apoptotic marker BCL-2, and apoptotic pathways are controlled by up-regulating pro-apoptotic proteins which are responsible for the cell death, such as, BAX and caspase 3, with an increase in DNA fragmentation. The pleotropic mechanisms of resveratrol are also accentuated by induction in the signaling pathways such as AMPK, SIRT1, HO-1, p53, p21, p16, eNOS and GSH-Px. NF-κB: nuclear factor-κB; MAPK: mitogen-activated protein kinase; TNF-α: tumor necrosis factor-α
Jang et al.[36] demonstrated an anti-inflammatory response in a rat model of inflammation by treatment with resveratrol, and further demonstrated reduction of prostaglandin synthesis by inhibition of cyclooxygenase-1 (COX-1). Later, it was found that resveratrol could selectively reduce COX activity by suppressing the COX-1 pathway; however, not through the COX-2 pathway. Szewczuk et al.[52] verified this observation. However, in yet another study, it was found that resveratrol inhibits the synthesis of prostaglandin E2 by suppression of COX-2, but not by altering COX-1[53]. One more study corroborates this result where resveratrol reduced colonic injury, neutrophil infiltration, and prostaglandin D2 concentration by inhibiting COX-2 without affecting COX-1[54]. It was also shown that resveratrol attenuates COX-2 expression[53].
Chemopreventive mechanism of resveratrol
Many studies have been published describing the potential chemopreventive effect of resveratrol. In brief, resveratrol induces apoptosis by interacting with the αVβ3 integrin receptor in the breast cancer cell line MCF-7[55]. The compound inhibits aryl hydrocarbon receptor activity thereby suppressing tumor growth and exhibiting anti-cancer properties[56]. Further, with MCF-7 cells, resveratrol inhibits NF-κB and BCL-2[57]. It antagonizes the aryl hydrocarbon receptor, which can relate to carcinogenic and immunosuppressive effects in cells[58]. Some studies suggest that resveratrol shows antitumor effects at the level of initiation, promotion, and progression with prostate cancer cells[59]. As noted above, resveratrol can inhibit COX-1 and COX-2, enzymes that are involved in tumor progression. With HL-60 cells, phenotypic markers indicative of reduced proliferation are induced. Also, in the initiation phase of carcinogenesis, resveratrol inhibits free radical generation[36]. In human lymphoblast cell lines, resveratrol induces apoptosis through p53 activation[60]. It also inhibits COX-2 activity[61] and ribonuclease reductase[62]. With osteosarcoma stem cells, resveratrol inhibits self-renewal, cell viability and tumorigenesis. Mechanically, resveratrol inhibited JAK2/STAT signaling and suppressed cytokine synthesis, which was consistent with the decline of CD133 cancer stem cell (CSC) markers[63]. Thus, when evaluated with in vitro models, resveratrol mediates a variety of effects consistent with antitumor activity.
Epigallocatechin-3-gallate
Chemical and pharmacological properties
Tea as a beverage is popularly consumed around the world, second only to water[64]. Consumption of green tea has been advocated for several health benefits, such as ameliorating cardiovascular risk and prevention of cancer. It has exhibited various biological properties proven favorable for hepatic function and improved metabolic profiles[65,66]. Pharmacologically, the polyphenols present in green tea demonstrate properties that can lead to anti-oxidative, anti-inflammatory, anti-atherosclerosis, anti-hypercholesterolemic and anti-carcinogenic activities[67,68].
Active green tea polyphenols include catechins (members of the flavonoid family), like epicatechin (EC), epicatechin-3-gallate (ECG), epigallocatechin (EGC), and epigallocatechin-3-gallate (EGCG). ECG and EGCG are found in the high concentrations (50 to 80% of the catechins)[64,69]. EGCG is the ester of epigallocatechin and gallic acid and known to be the most potent catechin in exhibiting anti-cancer and antioxidant effects. The hydroxyl group on the B-ring plays an important role in antioxidant reactions and this is increased by the trihydroxyl group on the D-ring[70].
Inflammation is one dominant factor in the initiation of cancer. Increased oxidative stress promotes cell growth. Many in vitro studies and studies with animal models have conclusively demonstrated the potential of green tea to reduce tumorigenesis, although clinical trials are still not definitive. EGCG is hydrolyzed to EGC in the intestine by bacteria. Later, EGC and gallic acid undergo several conversions leading to metabolites such as 5-(3,5-dihydroxyphenyl)-4-hydroxyvaleric acid and 5-(3ʹ,5ʹ-dihydroxyphenyl)-γ-valerolactone in glucuronide forms[71]. In a clinical study, the plasma levels were 0.17 µmol/L after having 2 cups of tea, while the in vitro concentrations used in many studies have been in the range of 10-100 µmol/L[66]. Epidemiological studies have suggested green tea has a positive effect on cancer prevention in certain types of cancer, namely breast, colon, and skin[72-74]. Although there is lesser or no positive clinical effect seen with other types of gastrointestinal or oral cancers, there is bactericidal activity against Escherichia coli, Streptococcus salivarius, and Streptococcus mutans[75-77].
Chemoprevention mechanism of EGCG
Since it has been established that cancer stem cells play a vital role in tumor progression, studies have been performed with lung CSCs such as A549 and H1299 illustrating the potential of EGCG to suppress tumor formation through the Wnt-β-catenin pathway[78]. Other studies performed with chemotherapeutic agent-resistant cancer stem cells such as A549/CDDP (cisplatin-resistant cells) and 5-flurouracil-resistant colorectal cancer cells have demonstrated the anti-cancer activity of EGCG[79,80]. Mechanistic evaluations with various in vitro models have led to the recognition of MAPKs as an important molecular target for EGCG [Figure 3]. These factors are associated with cell proliferation, differentiation, migration, senescence, and apoptosis[81]. ECGC inhibits signal regulated protein kinase and p38 phosphorylation. EGCG is also associated with inducing apoptosis, inhibition of transcription factors like NF-κB and activator protein (AP-1), and reduction of receptor tyrosine kinase activity[82].
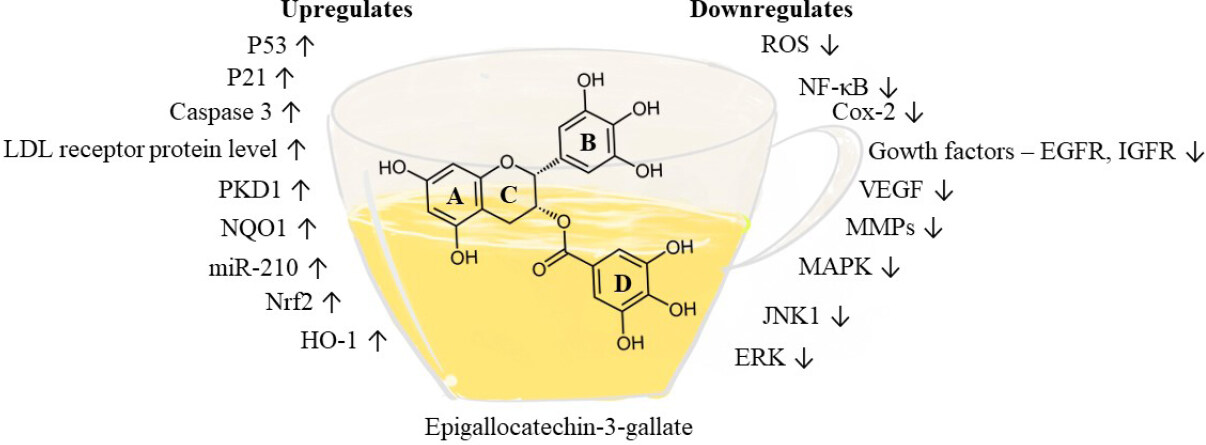
Figure 3. Pharmacological targets of epicatechin-3-gallate. EGCG affects various targets relevant to cancer prevention as shown in the figure. EGCG attenuates oxidative stress by down-regulating reactive oxygen species; inflammatory factors such as NF-κB and COX-2 are down-regulated. In addition, EGCG down-regulates several kinases, such as MAPK, JNK1, and ERK, and targets several growth factors, such as IGF1R, HGFR, and VEGF, that abate tumor growth and progression. Apoptosis factors such as p53, p21 and caspase 3 are up-regulated by EGCG, and the expression of NAD(P)H:quinone oxidoreductase 1 (NQO1), HO-1, and Nrf2 are increased to reduce oxidative stress. EGCG: epigallocatechin-3-gallate; NF-κB: nuclear factor-κB; COX: cyclooxygenase; MAPK: mitogen-activated protein kinase; ERK: extracellular signaling regulated kinases; VEGF: vascular endothelial growth factor
ERK signaling
ERK is one of the major signaling cascades of the MAPK signaling pathway that is targeted by EGCG. EGCG inhibits ERK activation in a concentration-dependent manner, suggesting its effectiveness as an anti-cancer agent with several in vitro models. With MCF10A and MDA-MB-231 breast cancer cells, 5 µmol/L EGCG inhibited hepatocyte growth factor-induced activation of ERK and AKT[83]. In cervical tumor cell lines, like HeLa, Caski and SiHa, EGCG inhibited phosphorylated ERK1/2 by 83% and Akt by 50% at a concentration of 50 µmol/L[84].
Activation of nuclear factor-κB and activator protein signaling pathways
Nuclear factor-κB (NF-κB) plays a vital role in the regulation of several genes central for cellular responses like inflammation, growth, and cell death. It is sequestered in cytoplasm in an inactive form and activated on phosphorylation. AP-1, a transcription factor, is known to be involved in tumor promotion and progression of cancer[85]. EGCG exerts inhibitory effects on the binding of NF-κB to DNA and thereby reduces inflammation and cell proliferation with in vitro models. However, concentrations required to mediate these effects were between 10-100 µmol/L[86,87]. EGCG evidently reduces binding of AP-1 along with NF-κB to DNA; a process which promotes MMP-9 for tumor progression. This has been demonstrated with several in vitro studies using human breast cancer cells[88], gastric AGS cells[89], and bladder cancer cells[90], in a dose-dependent manner, with concentrations ranging from 10 to 50 µmol/L.
c-Jun N-terminal kinase 1/2
JNK is involved in cancer cell apoptosis, although recent studies have indicated that Janus signaling promotes cancer cell survival by acting synergistically with NF-κB, JAK/STAT, and other signaling molecules. The situation is complicated because JNK1 and JNK2 have opposite effects in relation to cancer cell survival. JNK1 promotes apoptosis while JNK 2 promotes cancer cell survival. The expression of p53 is negatively regulated by JNK1 and positively regulated by JNK2[91]. EGCG attenuated reduced expression of JNK1 and oxidative damage and, at the same time, inhibited JNK2, thereby augmenting apoptotic signaling in cancer cells[87,92-94].
p38/MAPK signaling pathway.
p38/MAPK is a third major signaling cascade in MAPK, playing a major role in controlling the process of apoptosis. Activation occurs by several environmental factors such as stress and inflammatory cytokines. p38 activates several downstream kinases that induce apoptosis[95]. EGCG increases p38 levels and thereby inhibits growth in leukemic cells, HCC cells, and U373MG cells[94,96,97].
Sulforaphane
Chemical and pharmacological properties
Sulforaphane (1-isothiocyanato-4-methylsulfinylbutane) is an aliphatic hydrocarbon that is the major by-product obtained during the hydrolysis of glucoraphanin[98]. It was isolated in 1947 from radish. Later, glucoraphanin was found to be present in larger quantities in cruciferous vegetables such as cauliflower, broccoli, Brussel sprouts, and cabbage[99]. It is generally found in broccoli and broccoli sprouts, yielding the highest concentrations found in any plant source[100]. Broccoli and other cruciferous vegetables are widely consumed throughout the world for various health benefits.
Hydrolysis of glucoraphanin occurs due to disruption of the plant cell and the subsequent activity of the intrinsic enzyme myrosinase. Sulforaphane is not heat stable, but greater stability is retained when exposed to light and acidic pH levels, rendering the compound useful under gastric pH conditions[99-101]. The presence of epithiospecifier protein (ESP) disrupts the process of glucoraphanin hydrolysis, reducing the bioavailability of sulforaphane and sulforaphane nitrile, with the nitrile form being less active in its binding to pharmacological targets[102]. Since ESP is temperature insensitive, heating the broccoli at 60 ºC decreases the formation of sulforaphane nitrile[103]. Once absorbed, sulforaphane is conjugated with glutathione and metabolized by the meracaptopurine pathway; it is then excreted as N-acetylcysteine conjugates[104]. Pharmacokinetic studies have demonstrated that the peak plasma levels of sulforaphane are relatively high after oral administration of broccoli, for 1.6 to 6 h. with 95% elimination after 12 h[105]. In clinical studies, the plasma concentrations of sulforaphane following oral consumption of broccoli are in the range of 0.02-0.2 µmol/L[106]. With animal models, the consumption of broccoli has been found to exhibit protection against cancer[107].
Sulforaphane has also exhibited protective properties in the central nervous system by activating nuclear factor (erythroid derivative)-like 2 (Nrf2) and reducing oxidative stress and inflammation in nerve cells[108]. Further, the compound shows insulin-sensitizing and hepatoprotective effects in rats fed a high fructose diet[109]. Patients with type 2 diabetes treated with broccoli sprout powder (5 to 10 g for 4 weeks) showed reduced serum glucose levels and improved insulin levels[110]. Patients with deregulated type 2 diabetes treated with 5 g of broccoli extract along with metformin (500 mg to 3000 mg) showed reduced HbA1c levels and there was a reduction in glucose production[111]. In healthy male individuals, cholesterol and LDL cholesterol levels were reduced; in women, HDL cholesterol levels increased significantly after consumption of 100 g of fresh broccoli sprouts for 1 week[112].
Sulforaphane reduced levels of iNOS with lipopolysaccharide (LPS) activated macrophages in a mouse model[113]. Anti-inflammatory properties were further demonstrated in another study conducted with mice treated with sulforaphane; cytokine production was reduced in a concentration-dependent manner by activating the Nrf2 pathway. T-cell proliferation was also significantly inhibited[114].
Sulforaphane demonstrates inhibition of phase I and phase II enzymes, induces cell-cycle arrest and inhibits angiogenesis. At 15 µmol/L, sulforaphane promotes apoptosis and cell-cycle arrest in prostate cancer cells (LNCaP and PC3) by decreasing histone deacetylase (HDAC) enzymes[115].
Carcinogen-treated Wistar rats at 10 weeks of age were treated with 150 µmol of sulforaphane by oral gavage, mammary glands were extracted, and sulforaphane concentration after 12 h was 22 µmol/L. In addition, there was an increase of NQO1 and HO-1 levels observed in rat mammary gland. Subsequently, healthy women were placed on a cruciferous-free diet and administered 200 µmol of sulforaphane. The amount of sulforaphane distributed in breast tissue was found to be 0.92 ± 0.72 µmol/L[116].
It is documented in various epidemiological studies that the consumption of isothiocyanates from cruciferous vegetables is inversely proportional to the incidence of lung cancer cases[117]. This inverse correlation was even stronger in a study conducted in female patients who do not smoke cigarettes[118]. There is also substantial evidence based on studies conducted with cisplatin-resistant cancer stem cells [such as human non-small cell lung cancer (NSCLC)] that up-regulation of miR-214 induced by sulforaphane may lead to anti-cancer activity[119].
Chemopreventive mechanisms of sulforaphane
KEAP-nrf2 signaling
Genetic deletion of nrf2 can lead to detrimental effects on the survival of mice; they are more prone to brain injury and lung injury and other pathological conditions involving inflammation. To the contrary, the activation of KEAP1-nrf2 leads to protective effects in various animal models[120,121]. Nrf2 is important for the regulation of antioxidant genes such as enzymes that produce glutathione (GSH) and NADPH[122]. Sulforaphane, on entering cells, reacts with Kelch-like ECH associated protein, which functions as a sensor protein complex. Under basal conditions, KEAP1 binding to nrf1 leads to ubiquitination and proteasomal degradation. Sulforaphane protects nrf2 from degradation [Figure 4], allowing escape and the regulation of downstream target genes capable of mediating anti-inflammatory and antioxidant activities[123]. NQO1 and GST levels are significantly elevated in sulforaphane-treated wild-type mice (nrf2+/+) whereas nrf2-/- deficient mice exhibited no changes in NQO1[124].
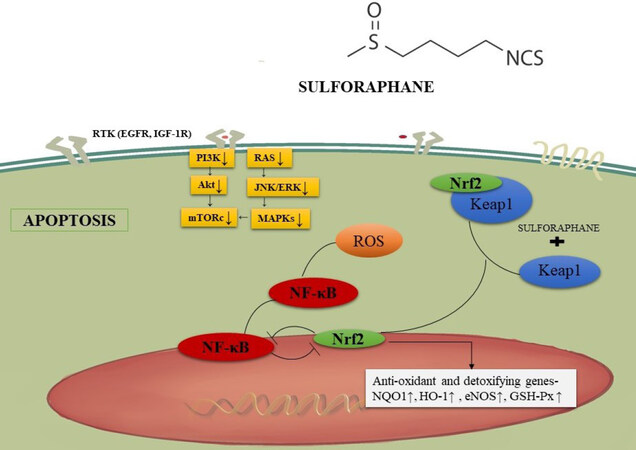
Figure 4. Sulforaphane and KEAP-nrf2 signaling. Sulforaphane enters cells and interacts with Kelch-like ECH associated protein. This prevents nrf2 degradation and leads to downstream anti-inflammatory and antioxidant effects. NF-κB: nuclear factor-κB
Activity of cyclin dependent kinase and reduced cyclin D1
Cyclin D1 is a cell-cycle regulator and a transcriptional modulator for histone deacetylase 3. Overexpression of these factors has been linked to cancer progression. Therefore, reduction of cyclin D is considered as a potential strategy for chemoprevention[125]. In this context, sulforaphane-treated A549 cells showed concentration-dependent reduction of cyclin D1 as well as increased expression of the p21[126]. In DU-145 prostate cancer cells, sulforaphane reduced CDK4 activity and cyclin D1 levels when treated with 9 and 50 μmol/L, respectively. CDK4 activity was also affected by a concentration of less than 1 μmol/L, but not significantly[127]. In an in vivo study, sulforaphane reduced tumor promotion and polyp formation in an ApcMin/+ mouse small intestine cancer model in a dose-dependent manner. However, biomarkers including cyclin D1 remained unaffected[128].
Inhibition of HDAC activity in human prostate and colorectal cancer cells
Increased HDAC expression has been associated with the deregulation of cell-cycle and apoptotic processes. HDAC inhibitors have shown potential in clinical studies for chemoprevention. Sulforaphane has demonstrated potential to reduce HDAC activity in prostate and colorectal cancer cells[129]. Sulforaphane (15 μmol/L) reduced HDAC activity by 30%, 40%, and 40% in LNCaP, BPH-1, and PC-3 cell lines, respectively. Expression of p21 associated with histone H4 was increased in all three cells lines leading to apoptosis[130]. In 4-6 week-old NOD/SCID mice inoculated with A549 lung cancer cells, administration of 9 μmol sulforaphane by oral gavage for 4 weeks attenuated the increase of tumor volume, significantly reduced HDAC activity, and increased acetylated histones 3 and 4[131]. HDAC inhibitory effects of sulforaphane were also demonstrated with a mouse model bearing a colon cancer xenograft[132]. The United States Food and Drug Administration approved the use of HDAC inhibitors for the treatment of cancer, so investigation of phytochemicals such as sulforaphane is reasonable[133]. In a phase II clinical trial involving patients with recurrent prostate cancer, administration of 200 μmol/day of sulforaphane rich extracts for 20 weeks did not produce a reduction in prostate specific antigen (PSA) levels by at least 50%. However, PSA doubling time was increased[134]. In a double-blinded study, there was a significant reduction in PSA levels in prostate cancer patients post-prostatectomy when given 60 mg sulforaphane orally with cancer therapy, followed by 2 months of sulforaphane with no other treatment[135]. The promising results observed with sulforaphane in these clinical trials suggest the use of an HDAC inhibitor in combination with a chemopreventive agent for the treatment of prostate cancer.
Anthocyanins
Chemical properties of anthocyanins
Anthocyanins are water-soluble secondary polyphenolic metabolites produced by plants[136]. The substances are classified under the flavonoid group and provide blue, red, and purple pigmentation for plants. The basic chemical structure of an anthocyanin contains anthocyanidin without a sugar moiety. The anthocyanidins are comprised of an aromatic ring (A) which is bonded to a heterocyclic ring (C) that includes oxygen, and this is similarly attached to carbon-carbon bonds linking to a third aromatic ring (B)[137]. There is a vast number of anthocyanins present in nature, the most common being petunidin, pelargonidin, delphinidin, peonidin, malvidin, and cyanidin. The only difference between these compounds is the nature and the number of sugars attached to their structure, the number of hydroxylated groups in the molecule, the aromatic or aliphatic carboxylates attached to the sugar, and the position of these bonds[138]. Studies have indicated the ortho-dihydroxyphenyl structure is the active moiety on the B-ring that suppresses tumor growth and metastasis[139,140].
Chemopreventive mechanism of anthocyanins
MAPK pathway and AP-1
With mouse epidermal cells (JB6+), tumor promoters such as epidermal growth factor (EGF), 12-O-tetradecanoylphorbol-13-acetate (TPA), and TNF-α can stimulate AP-1 activity and neoplastic transformation by inducing MAPK comprising ERK, p38 kinase, or JNK[141,142]. Cyanidin, petunidin, and delphinidin suppress TPA-stimulated AP-1 transcriptional activity and cell transformation with JB6+ cells[139].
Evaluation of structure-activity relationships have shown that the ortho-dihydroxyphenyl functional group on the B-ring of anthocyanidins plays a key role in inhibitory action since malvidin, pelargonidin, and peonidin, devoid of the ortho-dihydroxyphenyl structure, fail to block AP-1 activity or cell transformation. Signal transduction analysis showed that delphinidin blocked ERK phosphorylation at early times and JNK phosphorylation at later times, but p38 was not blocked [Figure 5][139]. Furthermore, delphinidin inhibits the phosphorylation of c-Jun (a phosphorylation target of JNK and ERK), SAPK/ERK kinase (SEK, a JNK kinase), and MAPK/ERK kinase (MEK, an ERK kinase). Suppression of TPA-induced AP-1 activity and cell transformation by delphinidin involves inhibition of JNK and ERK signaling cascades. This effect is increased in combination with superoxide dismutase (SOD) with any anthocyanidins bearing the ortho-dihydroxyphenyl arrangement on the B-ring. Multiplicative analysis demonstrated that inhibition between delphinidin and SOD is synergistic in nature[139]. Therefore, even though the signaling pathways affected by SOD or delphinidin are different, both are believed to play a crucial role in the cancer preventive activity of anthocyanidins.
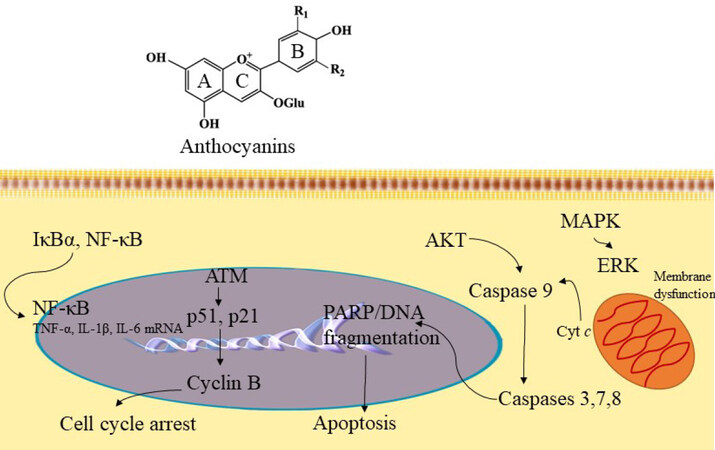
Figure 5. Anthocyanin mechanism of action. Based on structural-activity relationships (SAR), the ortho-dihydroxyphenyl functional group on the B-ring of anthocyanidins plays a key role in inhibiting the action of AP-1 and cell transformation. The compound inhibits MAPK/ERK and mitochondrial associated pathway caspases 3, 7, 8 to induce apoptosis. Further, anthocyanins have exhibited inhibition of cyclin-B by acting on p51 and p21 signaling pathways to induce cell cycle arrest. In addition, anthocyanins can act through the NF-κB signaling pathway to mediate anti-inflammatory functions. NF-κB: nuclear factor-κB; MAPK: mitogen-activated protein kinase; ERK: extracellular signaling regulated kinases
Suppression of inflammation through NF-κB and COX-2
There are some antioxidants that block the expression of COX-2 by inhibiting the signaling mechanism that controls the COX-2 gene[143]. In one study, the molecular mechanism of anthocyanins was evaluated using the mouse macrophage cell line RAW264. Bilberry and purified delphinidin derived anthocyanin extracts suppress LPS-induced COX-2 expression at the transcription and protein levels. Signal pathway analysis showed that delphinidin blocks LPS-induced IκB degradation and inhibits NF-κB stimulation and COX-2 gene expression. Thus, it appears anthocyanins act through the NF-κB signaling pathway which is involved in the suppression of COX-2 gene expression[144].
Apoptotic induction of cancer cells (by targeting ROS and JNK mediated caspase activation)
Petunidin, delphinidin, and cyanidin induced apoptosis with HL-60 cells, as demonstrated by DNA fragmentation and morphological changes, although peonidin, malvidin, and pelargonidin did not induce this response[145]. The number of hydroxyl groups at the B-ring is proportional to the potency of apoptosis induced by anthocyanidins; the ortho-dihydroxyphenyl structure at the B-ring is vital for apoptosis induction[145].
Mechanistic analysis indicated the induction of apoptosis by delphinidin may involve an oxidation/JNK-mediated caspase pathway. Delphinidin increases intracellular ROS which may trigger JNK. With delphinidin treated cells, JNK phosphorylation, caspase-3 activation, and c-jun gene expression were observed[145]. Delphinidin-induced JNK phosphorylation, DNA fragmentation, and caspase-3 activation were effectively blocked by antioxidants such as N-acetyl-L-cysteine[145]. Thus, delphinidin can potentiate the apoptotic death program in HL-60 cells through JNK signaling mediated through oxidative stress.
Chemopreventive mechanisms of anthocyanins
Anti-carcinogenic activities at the initial stage of tumorigenesis
Anthocyanins can prevent the occurrence of tumors by acting on antioxidant systems[146]. These compounds scavenge free radicals reducing damage to the genome of the normal cells by oxidative stress and later malignant transformation fostered by gene mutations[147,148]. Anthocyanins can induce the antioxidant effect by acting on the antioxidant response element through KEAP1-Nrf2. Furthermore, by regulating the expression of phase II antioxidases (quinone oxidoreductase, glutathione transferase, glutathione peroxidase, and glutathione reductase), anthocyanins can suppress the activity of caspase-3. The abnormal secretion and overexpression of inflammatory elements are important for tumorigenesis. It has been reported that anthocyanins can mediate anti-inflammatory functions by regulating the secretion and expression of inflammatory agents. This effect occurs due to the suppression of transcription factors NF-κB through multiple pathways[149,150]. In an in vitro study, anthocyanin containing purple-fleshed potato extracts elevated apoptosis and suppressed proliferation in a p53-independent manner in colon CSCs[151]. In vivo, purple-fleshed potato decreased the number of crypts containing cells with nuclear β-catenin (which is an indicator of colon CSCs) by induction of apoptosis[151]. These results provide evidence that anthocyanins may reduce the initial stage of tumorigenesis.
Anti-carcinogenic activities in the cancer formation stage
Anthocyanins can block tumorigenesis and induce terminal differentiation of tumor cells. It was found that differentiation of the acute promyelocytic leukemia cell line HL-60 could be induced by cyanidin-3-O-β-glucopyranoside (Cy-g) in a dose-dependent manner by the activation of PKC and PI3K. Upon treating HL-60 cells with Cy-g (200 µg/mL), differentiation characteristics were observed such as enhanced activity of esterase, increased adhesion, and reduced expression of the oncogene c-Myc. When cells were treated with PKC or PI3K inhibitors, the effect of differentiation induced by Cy-g was substantially decreased[152]. Since the degree of differentiation correlates with the degree of tumor malignancy, it may be suggested anthocyanins can act at the cancer formation stage by stimulating differentiation.
Anti-carcinogenic activities in the cancer development stage
Anthocyanins can induce apoptosis of cancer cells through the external death receptor pathway and the internal mitochondrial pathway. It was determined that delphinidin could activate p38-FasL and the Bid pathway, which is a pro-apoptosis protein that induces apoptosis with HL-60 cells, in a dose- and time-dependent manner[153]. In vascular smooth muscle cells, delphinidin and cyanidin strongly inhibited the expression of vascular endothelial growth factor (VEGF) (stimulated by platelet derived growth factor) by repressing the JNK and p38-MAPK pathways[154].
Pharmacological properties
Hydroxylation of nonreactive carbons is a major function of phase I cytochrome P450s and the mono-oxygenase system[155]. Phase I hydroxylation of anthocyanins is noteworthy since hydroxyl groups structurally distinguish this group of compounds. Even though flavonoids are reported to have low bioavailability due to extensive metabolism, their metabolites may be present for a longer duration and result in significant bioactivity. This can be the case with anthocyanins, since metabolites have been found to retain basic structural characteristics, thereby preserving bioactivity[156,157]. The majority of the flavonoids present in the urine and circulation as glycated, glucuronidated, sulfated, and methylated conjugates[158,159]. Glucuronide conjugation is regarded as an important conjugation reaction in the metabolism of flavonoid[158-162]. UDP-glucuronosyltransferases, which catalyzes the glucuronidation reaction, are observed in high concentrations in the intestine, kidney, and liver[163,164]. However, in humans, following dietary consumption, evidence suggests that the initial site for flavonoid glucuronidation is the intestine[159,165,166]. Methylation is observed as the second major conjugation reaction of flavonoids[162]. O-Methylation is the most common such reaction. Catechol-O-methyltransferase catalyzes O-methylation utilizing the cofactor S-adenosyl methionine. The liver is a major organ responsible for methylation with the highest catechol-O-methyltransferase activity[167]. The primary site of methylation is determined by the pattern of a flavonoid ring structure. Some studies have shown low oral doses of quercetin in animals and humans undergo extensive methylation[168]. Glycation or sulfation are common conjugation reactions that predominate when low dose phenolic drugs are administered. Sulfotransferases are a small group of cytosolic enzymes that are widely distributed in the body. They use phosphoadenosine-5’-phosphosulfate as a cofactor and their recognized substrates include polyphenols (i.e., flavonoids), hydroxylamines, 4-nitrophenol, iodothyronines, and phenols[168].
Studies suggest that anthocyanins have very low bioavailability (< 1% in plasma), although some amount has been found in the colonic tissues of patients, indicating the possibility of a local site of action[169,170]. It is still unclear whether anthocyanins are effective against cancer in human beings and whether they can function as metabolites or as parent molecules.
Curcumin
Chemical and pharmacological properties
Curcumin [(1E,6E)-1,7-bis(4-hydroxy-3-methoxyphenyl)-1,6-heptadiene-3,5-dione] is a yellow-colored phytochemical obtained from Curcuma longa (turmeric), a member of the ginger family (Zingiberaceae). It is commonly cultivated in the southwest Asia region and is popularly used as a spice for various cuisines[171,172]. Curcumin is comprised of two o-methoxy phenolic rings which are connected by seven carbon chains consisting of an α, β-unsaturated β-diketone component, and generally accounts for 60%-70% of curcuminoid extracts[173,174]. The o-methoxyphenol rings are the primary domains for the antioxidant activity of curcumin and the β-diketone component forms chelation complexes with metals rendering them useful for the treatment of heavy metal poisoning[175].
In a pre-clinical study, 500 mg/kg curcumin administered orally to rats gave a maximum plasma concentration of approximately 0.06 µg/mL, reaching the peak by approximately 41 min; the half-life was 28 min, giving an oral bioavailability of about 1%[176]. As suggested by this poor bioavailability, the tolerance for curcumin is very high for humans. In a dose escalation study, minimal adverse events were observed which were unrelated to the dose after oral administration of 10,000 or 12,000 mg in healthy subjects. Still, only traces were detected in serum[177]. At 10-12 g, the highest dose given to human subjects, the plasma concentration of curcumin and its metabolites were in the range of 0.075-10 µg/mL[178]. Curcumin is metabolized by the process of glucuronidation in the intestine and by the liver yielding metabolites such as tetra-hydrocurcumin, and hexahydrocurcumin[179].
Nevertheless, there are a vast number of studies conducted with curcumin to investigate anti-inflammatory, antioxidant, anti-carcinogenic, antiviral, and anti-infection activities. Owing to its antibacterial, antioxidant, anti-inflammatory, and antiseptic properties, curcumin has been proven useful in treating several skin diseases, such as psoriasis, infection, acne, skin inflammation, and skin cancer[180]. Curcumin controlled the in vitro growth of Staphylococcus aureus and Pseudomonas aeruginosa in an in vivo murine wound model, and topical administration accelerated healing[181]. In a psoriasis keratin (K) 14-VEGF transgenic mouse model, after oral curcumin administration, symptoms like ear redness, weight, thickness, and lymph node weight were reduced significantly[182].
There have been several conclusive in vitro, in vivo, and clinical studies conducted on the use and benefits of curcumin in treating inflammatory diseases. Curcumin exhibited anti-inflammatory effects in adipose tissue of C57BL/6 mice fed a high fat diet by down-regulating inflammatory transcription factors like NF-κB and toll-like receptor-4[183]. The clinical effectiveness of curcumin in treating inflammation was studied in patients diagnosed with osteoarthritis, and significant reductions in myeloperoxidase, collagen degradation activity, and C-reactive protein levels were observed; also, additional symptoms like pain and effusion were reduced[184]. In 45 patients with active rheumatoid arthritis, curcumin (500 mg) was used in combination with diclofenac sodium (50 mg) and there was a significant reduction in their disease activity score compared with the group provided only with diclofenac sodium[185]. In yet another clinical study, patients with acute rhinitis were administered 500 mg of curcumin orally. Symptoms like sneezing, itching, and obstruction rhinorrhea were reduced significantly in patients treated with curcumin compared to the placebo treatment, and inflammatory markers such as IL-4, TNF-α, IL-8, PEG2 were significantly decreased[186].
Curcumin as a chemopreventive and anticarcinogenic agent
Curcumin has been broadly studied for anti-cancer characteristics [Figure 6]. A pilot study conducted with patients diagnosed with benign prostatic hyperplasia (BPH) and administered 500 mg of curcumin twice a day along with the standard therapeutic regimen showed improved symptoms and improved quality of life relative to the group administered the standard treatment only[187]. In another study, 85 patients with prostate cancer were administered curcumin and soy isoflavones along with the standard treatment, and elevated PSA levels were significantly decreased in patients treated with curcumin and soy isoflavones compared with the group given the standard treatment[188].
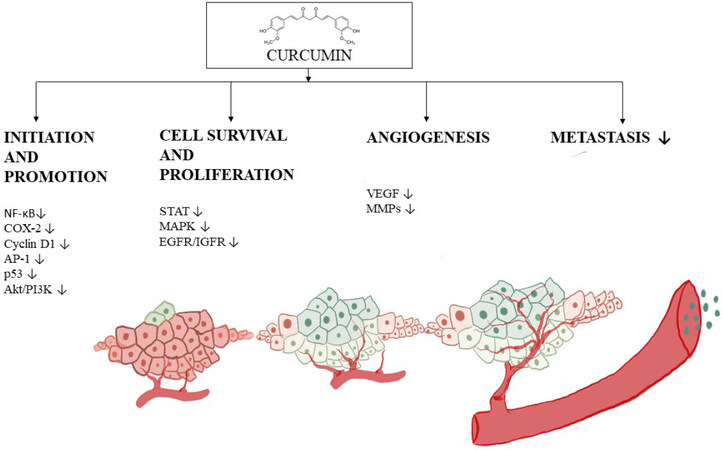
Figure 6. Progression of angiogenesis and the inhibitory mechanism of curcumin. Curcumin is capable of functioning through a myriad of mechanisms, but this figure illustrates down-regulation of factors involved in tumor initiation such as NF-κB, COX-2, cyclin D1, p53 and Akt/PI3k, and proliferation factors such as STAT, MAPK EGFR. The progression phase of tumorigenesis is also inhibited, for example, by alteration of VEGF expression, leading to reduced angiogenesis and metastasis. NF-κB: nuclear factor-κB; COX: cyclooxygenase; PI3K: phosphatidylinositol-3-kinase; MAPK: mitogen-activated protein kinase
In other forms of cancer, like breast and cervical, curcumin has proved effective. In patients with advanced metastatic breast cancer, curcumin reduced biomarkers like CEA with a maximum tolerated dose of 8000 mg along with chemotherapy[189]. In addition, topical application of curcumin along with other herbal preparations for 30 consecutive days resulted in significant clearance of HPV. However, vaginal curcumin capsules did not show any statistical difference[190]. Oral administration of curcumin (8000 mg) is well tolerated in patients diagnosed with advanced pancreatic cancer[191]. And, in a Phase II clinical trial, despite its limited absorption, curcumin exerted biological activity in a few patients, such as down-regulation of transcription factor NF-κB and reduced COX-2 (8,000 mg/day dosage)[192].
Mechanism of anti-carcinogenesis
VEGF
Since VEGF is a crucial regulator for angiogenesis, the use of potentially active phytochemicals including curcumin as an anti-angiogenic compound has been explored[193]. With the H22 HCC cell line, curcumin inhibited the proliferation of cells in vitro and reduced VEGF expression and PI3K/AKT signaling in vivo[194]. Curcumin reduced high mobility group box 1 and VEGF-D expression in gastric cancer AGC and SGC-7901 cell lines, and induced cell apoptosis through caspase-3 activation, in a dose-dependent manner[195].
Role of curcumin in cell-cycle arrest
Curcumin inhibits the expression of cyclin E and cyclin D1 that enable cell-cycle arrest at the G1/S phase in LNCaP and PC-3 cell lines[196]. In another in vitro study, with human osteosarcoma cells, cell-cycle arrest by curcumin at the G1/S phase was related to a reduction of cyclin D1[197]. Human mantle cell lymphoma is characterized by overexpression of cyclin D1, which up-regulates NF-κB and several genes such as, bcl-2, COX-2 and IL-6. However, on treatment with curcumin, cyclin D1 expression was down-regulated, along with AKT activation[198]. Curcumin also improves the efficacy of chemotherapeutic agents such as cisplatin by targeting cancer stem cells [such as A549 and H2170 (NSCLC cell lines)], down-regulating cyclin D1 and increasing the expression of p21[199]. There is also evidence of involvement of curcumin in suppressing glioma growth and inducing apoptosis by AKT and signal regulated kinase pathway in U87-MG cells[200]. In T-cell acute leukemia malignant cells, curcumin causes de-phosphorylation of active AKT, FOXO, and releases cytochrome c, which activates caspase-3. This leads to inhibition of cell proliferation[201].
Silibinin
Chemical properties of silibinin
Silibinin is a key constituent of silymarin, which is a polyphenolic flavonoid obtained from milk thistle seeds[202]. It is constituted as two isomers, silybin A and silybin B, in the ratio of 1:1, functions as a weak acid in aqueous form, and is especially stable in the presence of acids and unstable in a basic environment[203]. Silibinin is a major component of the silymarin complex, constituting 50%-60%, depending on the formulation[204]. Many studies have demonstrated that silibinin has strong antioxidant properties, scavenging both reactive oxygen species and free radicals, which can lead to the enhancement of cellular antioxidant defense mechanisms[205-209]. Notably, silibinin can affect cancer development by various modes of action, including modulation of oxidative stress, proliferation, inflammation, metastasis, and angiogenesis[210-212]. Some studies have indicated a beneficial effect on toxicity due to short- and long-term exposure to radiation treatment and chemotherapy[213].
Pharmacokinetics of silibinin
Silymarin is primarily conjugated and excreted into bile and urine and seems to undergo insignificant phase I metabolism; inadequate data exist concerning the role for phase II metabolism and transporters[214,215]. Silymarin (where silibinin is an active constitute) pharmacokinetic analysis was done with healthy volunteers. There was rapid metabolism, forming conjugates such as glucuronides, that were detected in plasma[214]. Also, it was found that conjugated silibinin metabolites are eliminated slowly as compared to free silibinin[214]. Factors such as inefficient intestinal absorption, low water solubility, elevated metabolism, and rapid excretion, significantly decrease the serum concentration of silibinin, thus reducing its ability to reach target organs and consequently therapeutic efficiency is reduced[216-218]. There have been many efforts to produce formulations to increase the bioavailability of silibinin[219,220]. For example, silibinin that is complexed with phosphatidylcholine is known as “silipide”. In pharmacokinetic studies conducted with healthy subjects, it was shown that silibinin derived from silipide has greater absorption in plasma and liver as compared with conventional silibinin[221,222]. Silipide, when tested in cancer patients, demonstrated high plasma bioavailability[223-225]. A comparative study revealed high bioavailability of silibinin in colon tissue but relatively poor levels in prostate tissue[223-225]. This suggests organ specificity may be anticipated as a result of bioavailability following oral administration.
Chemoprevention mechanism of silibinin
Several studies suggest that silibinin may be effective against lung cancer. Silibinin reduced the production of matrix metalloproteinase-2 and urokinase-plasminogen activator when metastatic A549 lung cancer cells were treated with different concentrations, up to 100 µmol/L[226]. Silibinin has been shown to reduce the development of human NSCLC, such as large cell carcinoma cells (H1299 and H460) and bronchioalveolar carcinoma cells (H322)[227]. Silibinin treatment of cultured cells (10-75 µmol/L) has been shown to target cell-cycle progression leading to a G1 arrest and altered protein levels of cyclins (D1, D3, and E), CDKIs (p18/INK4C, p21/Cip1, and p27/Kip1) and cyclin-dependent kinases (CDKs)[227]. Inhibitory effects of silibinin (75 µmol/L) were shown in both a human large-cell lung cancer cell line and human lung adenocarcinoma A549 cells[228]. A combination of silibinin and indole-3-carbinol showed greater anti-proliferative effects than the single compounds alone. A549 cells, when treated with 100 µmol/L of indole 3-carbinol and 75 µmol/L of silibinin, or 200 µmol/L of indole-3-carbinol plus 75 µmol/L of silibinin, for 24 h, showed a reduction in proliferation by 40% and 62%, respectively. With H460 cells, the responses were 31% and 69%, respectively. At the molecular level, the combination of indole-3-carbinol and silibinin inhibited Akt and ERK activation and induced apoptosis[228]. Silibinin has also shown effects against cancer stem cells (CSC) in lung cancer[229]. A model of acquired erlotinib resistance was established by growing NSCLC cells with a TKI-sensitizing EGFR exon 19 deletion in the constant presence of high doses of Erlotinib (Erlotinib-refractory PC-9/Erl-R cells). Treatment with silibinin (50-100 µg/mL) decreased the amount of lung cancer spheres in this model. These results suggest the possibility of using silibinin in combination with EGFR tyrosine kinase inhibitor Erlotinib to target CSC in EGFR-mutant NSCLC patients[229].
The anti-prostate cancer properties of silibinin have been investigated in various studies. The substance has shown inhibition of androgen- and serum-stimulated PSA protein levels in LNCaP cells, and this is associated with cell growth inhibition through G1 arrest in cell cycle progression[230]. Treatment with 25 and 75 µg/mL of silibinin for 24 h showed 45% and 59% decreases in PSA secretion in the medium, respectively. Silibinin has further shown up-regulation of insulin-like growth factor binding protein 3 expression and suppression of androgen-independent prostate cancer (PC-3) cell proliferation[231]. With PC-3 cells in a medium supplemented with 10% FBS, treatment with silibinin (2 and 20 µmol/L) reduced cell growth by 17.3% and 54%, respectively[231].
Silibinin has been shown to suppress the adhesion and migration of human prostate adenocarcinoma (PC-3) cells[232], and the motility, migration, and invasion of ARCaPM prostate cancer cells[233]. However, the response was weak with ARCaPM cells. When applied at a higher concentration (200 µmol/L), growth inhibition was only 18.5% in ARCaPM cells, but in the case of LNCaP, PC-3 and DU145 cells, the growth inhibition was found to be 48.7%, 60.0%, and 73.8%, respectively, after 48 hours of treatment. Moreover, silibinin induced morphological reversal to the epithelial phenotype from epithelial-mesenchymal transition (EMT), down-regulated MMP-2 and vimentin, and up-regulated cytokeratin-18. Also, silibinin inhibited NF-κB p50 translocation by up-regulating I kappa B alpha (IκBα) protein, and down-regulating the expression of two key EMT regulators, SLUG, and ZEB1 transcription factors[234].
Several studies have demonstrated the activity of silibinin against colon cancer. Silibinin mediated apoptosis in cultured human colorectal carcinoma LoVo cells, which was associated with high levels of cleaved PARP and cleaved caspases (3 and 9). When LoVo cells were treated with silibinin (50-200 µmol/L) for 24 hours, growth was reduced by 30%-49%. The substance also induced strong cell cycle arrest at the G1 phase, and a minor although significant G2/M phase arrest, at the highest concentration tested. Moreover, silibinin reduced the levels of cyclins (A, B1, D1 and D3), CDK1, CDK2, CDK4, and CDK6, and elevated the level of CDKIs (p21 and p27) and phosphorylation of Rb protein[235]. Anti-angiogenic effects, such as a decrease in inducible COX-1 and COX-2, NOS and NOS3, and VEGF and HIF-1α, were exhibited by silibinin[236]. When administered along with TNF-related apoptosis-inducing agent (TRAIL), silibinin caused cell death in primary tumor cells (SW480), as well as TRAIL-resistant metastatic cells (SW620). Finally, silibinin induced up-regulation of death receptor 4 (DR4) and DR5, down-regulated the anti-apoptotic proteins XIAP and Mcl-1, and synergistically activated the mitochondrial apoptotic pathway[237][Figure 7].
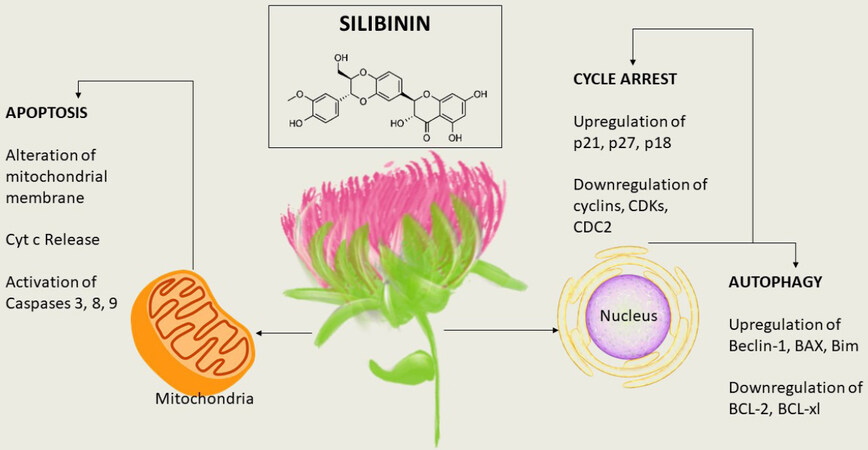
Figure 7. The molecular targets of silibinin. Silibinin exhibits a decrease in mitochondrial function and releases mitochondrial cytochrome c. The compound initiates cell death in primary tumor cells and causes up-regulation in p21, p27, and p18, leading to cell-cycle arrest. In addition, autophagy is induced by up-regulation of BAX, Bim, and Beclin-1
Lycopene
Chemical properties lycopene
Lycopene is a lipophilic hydrocarbon carotenoid with red color due to a chromatophore with 11 conjugated double bonds[238]. The antioxidant properties of lycopene derive from this conjugated polyene structure[239]. Unlike α- and β-carotenes, lycopene is not provitamin A due to the absence of a β-ionone ring and its acyclic structure[239]. Having been studied for many decades, lycopene is found in tomatoes, watermelon, pink grapefruit, papaya, and other fruits[240]. Unlike other widely distributed carotenoids, however, lycopene is mainly found in tomatoes and tomato products[241]. Unique structural and chemical features may contribute to distinct biological properties[239].
Pharmacological properties of lycopene
Lycopene has been shown to function through a variety of mechanisms [Figure 8], several of which are described below.
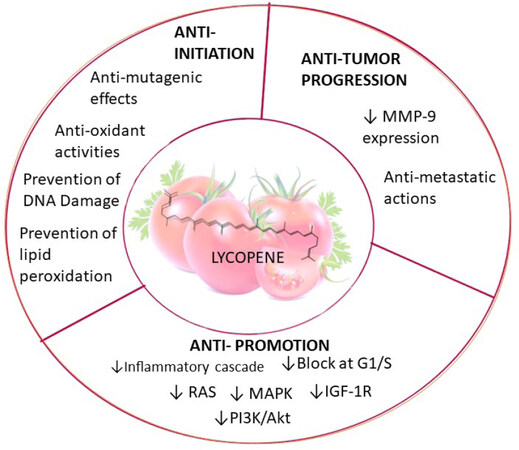
Figure 8. Pharmacological functions of lycoepene. Lycopene induces factors leading to anti-initiation and anti-progression by down-regulating MMP-9 expression. The compound mediates anti-promotion effects by suppressing RAS, MAPK, IGZF-1R, PI3K/AKT, and inflammatory cascades. MAPK: mitogen-activated protein kinase; PI3K: phosphatidylinositol-3-kinase
Antioxidant activity
Oxidative stress caused by the ROS is linked with aging, carcinogenesis, and cardiovascular diseases. Lycopene can function as an antioxidant by various mechanisms, with the best documented mechanism being through quenching singlet oxygen (1O2) due to the extended system of conjugated double bonds[242].
In cell culture, V79 Chinese hamster lung fibroblasts were treated with peroxynitrite to cause DNA strand breakage and nitration of proteins. This was shown to be inhibited by lycopene in the concentration range of 0.31 to 10 µmol/L[243]. In another preclinical study, lycopene at concentrations of 0.25 to 10 µmol/L, reduced the oxidative DNA damage caused by the redox-cycling of catechol estrogens in Chinese hamster lung fibroblasts and plasmid DNA[244]. It was determined that 81% of the subcellular localization of lycopene in prostate cancer cells treated with lycopene was at the nucleus[245]. Consistent with this, it is observed that the DNA protective effects exhibited by lycopene correlate with the localization of lycopene in the nucleus[246].
Apoptosis
In one study, lycopene reduced mitochondrial transmembrane potential, decreased mitochondrial function, increased annexin V binding, and released mitochondrial cytochrome c[247]. In another study, apoptosis was induced with LNCaP and PC3 cells with lycopene concentrations as low as 10 nmol/L[248].
With human colon carcinoma (HuCC) cells, lycopene treatment at 2.0 or 4.0 µmol/L induced apoptosis. However, the compound was not effective when incubated with a physiologically relevant concentration 1.0 µmol/L. The same group showed apoptosis in Raji cells, a prototype Burkitt lymphoma cell line, when exposed to 2.0 µmol/L lycopene. There were no anti-apoptotic effects observed in human erythroleukemia (K562) or B chronic lymphocytic leukemia (EHEB) cells treated with lycopene at a concentration of 4 µmol/L[249]. These data indicate that lycopene shows greatest efficacy for inducing apoptosis in human prostate cancer cells.
Cell-cycle arrest
Studies have suggested that lycopene can induce cell-cycle arrest at the G1 phase. It was found that Hep3B human hepatoma cells were inhibited 20%-50% by lycopene at physiologically relevant concentrations as low as 0.2 µmol/L. Lycopene induced G0/G1 arrest[246]. Similarly, in a study with human prostate cancer cell lines PC3 and LNCaP, lycopene promoted mitotic arrest at the G0/G1 phase, facilitated by a decreased amount of cyclin dependent kinase 4 and cyclin D1 and E[248].
Pharmacokinetics and bioavailability
The pharmacokinetics of lycopene provides insight regarding relevant mechanisms by which intervention impacts disease. The ability to interpret epidemiologic findings, design clinical intervention trials, and plan physiologically relevant animal and in vitro mechanistic investigations hinges on overall bioavailability. Critical questions impacting interventions include absorption, distribution, tissue concentrations and metabolic breakdown.
One important parameter is plasma half-life. With patients given 20 mg of lycopene from tomato soup or a synthetic lycopene tablet for 8 sequential days, the systemic availability of lycopene in the form of a tablet was comparable to that of processed tomatoes (tomato paste available as a soup) and better than tomato juice. The plasma lycopene half-life was found to be 5.6 and 6.4 d for the tablets and the soup, respectively. Furthermore, the half-lives of 5-cis- vs. all-trans-lycopene were evaluated and found to be 7.4 and 5.4 d, respectively. These observations suggest differences in the pharmacokinetic parameter which may lead to distinctive tissue and plasma lycopene isomeric patterns[250]. The difference in the half-lives observed in the geometrical isomers of lycopene may be due to factors such as the higher bioavailability of cis vs. trans[251,252], greater thermodynamic stability of cis isomers at elevated temperature[253], and endogenous isomerization, which may or may not be enzymatic[254].
Cleavage of β-carotene gives homologous carbonyl products[255,256]; the pathway was confirmed by the identification of BCO2 in zebra fish, ferrets, mice and humans[257,258]. BCO2, an enzyme derived from mitochondria[259], is mainly expressed in testis and liver, with a comparatively lower amount found in lung, heart, brain, colon, stomach, intestine, prostate, spleen, and kidney[257,258]. In a study conducted with humans with lycopene, plasma of those who consumed tomato juice was found to contain apo-6-, apo-8’-, apo-10’-, apo-12’-, and apo-14’-lycopenal[260]. Moreover, it was reported that non-provitamin A carotenoids, including zeaxanthin and lutein, are preferentially cleaved relative to provitamin A carotenoids, which indicates a major role of BCO2 in the metabolism of non-provitamin A carotenoid[258,261]. In another investigation with humans given lycopene, a series of apo-lycopenals were identified in plasma, including apo-10’-lycopenal[260]. However, it is not known whether these metabolites are enzymatically cleaved or products originating due to chemical oxidative cleavage[260].
In a single blind, randomized, placebo-controlled study, lycopene supplements of 15 mg two times a day for 3 weeks (Lyc-O-Mato™, LycoRed, Beer-Sheva, Israel) were given to 15 of 26 men who were scheduled for radical prostatectomy due to organ confined malignancy. Over 3 weeks, in those taking lycopene supplements, a 22% increase in tissue and plasma levels and a statistically significant reduction in PSA was observed. Tumor volume was reduced. In addition, cellular proliferation biomarkers were decreased whereas cellular differentiation markers including connexin 43 and apoptosis were increased with intervention[262]. In another study, it was reported that combined intake of tomato sauce, tomatoes, tomato juice, and pizza decreased the risk of prostate cancer[263]. These findings suggest that tomato-based foods may be beneficial for prostate cancer.
Conclusions and future directions
We currently review some well-studied phytochemicals with cancer chemopreventive potential. Of course, many other cancer chemopreventive agents are known, either naturally occurring or synthetic, such as genistein[264], 6-gingerol[265], diallyl disulfide[266], lupeol[267], honokiol[268], plumbagin[269], ellagic acid[270], and quercetin[271], but our arbitrary selection mainly focuses on agents found in the diet with strong evidence of bioactivity. Numerous studies have been completed or are ongoing with these compounds; copious amounts of data have been generated. As one example, more than 20,000 articles were disclosed when searching “resveratrol” as a topic on Web of Science (accessed on August 25, 2020), and there is no doubt a search involving other chemopreventive agents would reveal a multitude of publications as well. Given this huge corpus of data, and taking into account the current state of science, there is now an opportunity to further expand the field of chemoprevention. We currently suggest three such paradigm shifts, referred to as omics, indirectness, and sequelae.
Omics
The traditional dogma for the discovery of drug candidates is based on “one drug-one target”, where the goal is to find highly selective agents with one molecular target (or biomarker). The concept is to minimize adverse effects caused by off-target responses. However, this approach is disingenuous. First of all, an agent functioning in such a manner would be highly susceptible to failure due to the possibility of developing drug resistance. Moreover, signaling pathways are exceedingly linked via interconnected networks (cf. Figure 9). A small change in one target can result in multiple changes in other pathways, akin to the so-called Butterfly Effect (“Does the Flap of a Butterfly’s Wings in Brazil Set Off a Tornado in Texas?”)[272]; Also, the genesis of cancer is comprised of multiple factors, not a single specific element. This is further complicated by a host of endogenous (e.g., aging, genetic susceptibility, DNA repair machinery, hormones, growth factors, inflammation) and exogenous (e.g., radiation, chemical carcinogens, viruses, smoking, lack of exercise, nutrient imbalance) risk factors, and typically long latency periods[273]. Further, cause-effect relationships are often nebulous.
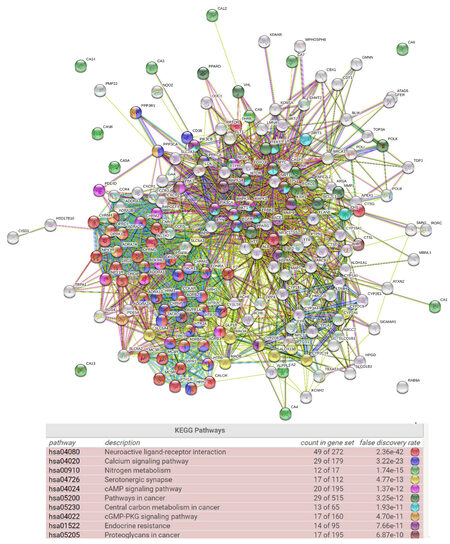
Figure 9. Bubble map with target proteins of resveratrol using STRING software. Human target proteins of resveratrol that were selected from ChEMBL are visualized as a network of predicted associations. The network nodes represent proteins while the edges represent the protein-protein associations. The colored lines represent different types of evidence - Light blue line: curated database; purple line: experimentally determined; green line: neighborhood; red line: gene fusion; blue line: gene co-occurrence; yellow line: text mining; black line: co-expression. The colored nodes demonstrate the pathways. The KEGG pathway table demonstrates the top ten pathways enriched by resveratrol based on a gene list of human target proteins. Neuroactive ligand-receptor interaction appears at the top of the list with the lowest false discovery rate (2.36 × 1042)
In the context of chemoprevention, it is likely that dietary phytochemicals currently play an important role in cancer reduction or delay, but that role is difficult to quantify in a direct manner. Daily consumption varies, as does the phytochemical content of the foods consumed. For a more predictable response, especially for individuals at high risk for developing malignancies, a cocktail of chemopreventive agents would be preferred[8]. Development of such a cocktail needs to take into account the pleiotropic activities of typical chemopreventive agents, as demonstrated by those described in this review and elsewhere. On one hand, pleiotropic responses are considered a distinct advantage. On the other hand, the creation of a proper preparation becomes a daunting task as a result of complexity. However, unique tools are now available that can be put to use. For example, with agents know to function in a chemopreventive capacity, we can take into account novel pathways uncovered utilizing primary -omics data (e.g., genomics, proteomics, metabolomics) or data mining with publicly accessible biological data repositories (e.g., ChEMBL, PubChem).
Consider resveratrol as an example. Based on literature reports describing individual actions of this compound, we input human target proteins listed in ChEMBL (accessed on August 25 2020)[274] on STRING (accessed on August 25 2020)[275]. As shown in Figure 9, the gene list with network edges is visualized with enriched pathways (e.g., neuroactive ligand-receptor interaction, calcium signaling pathway, nitrogen metabolism, serotonergic synapse, cAMP signaling pathway). Clearly, this is a more realistic view of the action of resveratrol, relative to thinking of it as simply inhibiting or stimulating factor x, y or z. Now, if we consider a second chemopreventive agent, and the network of factors modulated by that agent, and overlay the two individual networks, it is easy to perceive the complexity of the actual response leading to a chemopreventive response.
There is little doubt that the intrinsic response of a mammal exposed to a chemopreventive agent correlates to some extent with the theoretical response shown in Figure 9. As an example, an area of great interest for our group is the potential effect of grapes on health[276], and the corresponding mechanisms. In a recent study (unpublished) in which mice were provided diets with or without whole grape supplementation, RNA expression data were examined. As shown in Figure 10, remarkable alterations were observed that would likely relate to some of the beneficial properties. There is little doubt this modulation of genetic expression results from the action of phytochemicals contained within the grape. Grapes have received great notoriety for being a primary dietary source of resveratrol but, in actual fact, whole grapes contain over 1,600 phytochemicals[34]. Thus, it is perhaps not surprising that such a profound effect on genetic expression was observed. This provides a good illustration of the complexity of a real-life response and accentuates the naivety of one drug-one target philosophy.
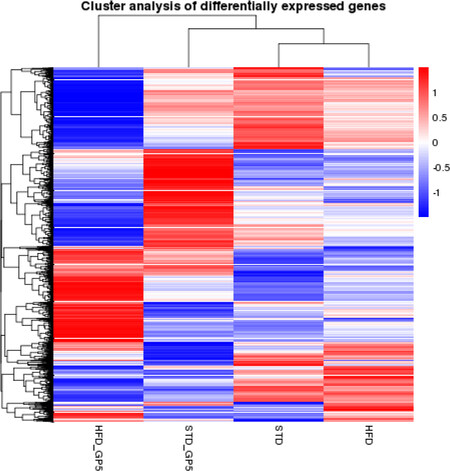
Figure 10. Hierarchical clustering heatmap. A study was conducted with female C57BL/6J mice. Starting at age 10 weeks, animals were provided the following diets ad libitum: standard synthetic diet (STD); STD supplemented with 5% whole grape powder (STD5GP); a high-fat (Western) diet (HFD); HFD supplemented with 5% whole grape powder (HFD5GP). Cluster analysis was produced using the log10(FPKM)+1 value that shows the influence of grape on the expression levels of genes. Red color indicates up-regulated gene expression; blue color indicates down-regulated gene expression. The interrelationships between groups is presented as hierarchical clustering. The order of the rows and columns is based on resemblance correlations that can be interpreted based on the hierarchical clustering shown in the heatmap. The data indicate dietary supplementation with whole grape powder influences the expression levels of genes. The heatmap was produced by analyzing a total of 11,047 genes
Indirectness
As noted above, the low bioavailability of phytochemicals may lead to discrepancies in effective doses observed with in vitro models but required for in vivo responses. For example, the oral bioavailability of resveratrol is reported as under 1% due to rapid metabolism in the intestine and liver[277]. As exemplified in Figure 11, this has led to the exploration of technology designed to enhance bioavailability. Here, a map was created based on bibliographic data (co-occurrence) of “resveratrol” from Web of Science, and words such as “drug-delivery” and “encapsulation”, and highlighting using VOSviewer[278].

Figure 11. Density visualization of “resveratrol” and co-occurring words by VOSviewer. Co-occurrence words with resveratrol are displayed based on text data from Web of Science. The color illustrated in the figure changes from blue to yellow based on the frequency with which as a word is linked to resveratrol
Of course, metabolism of chemopreventive agents may have a major influence on efficacy. Several major metabolites derived from the chemopreventive agents described herein are shown in Figure 12. In some cases, these metabolites may be less active than the parent molecule. In other cases, such as with resveratrol 3’-sulfate[279], the activity may be similar or even greater.
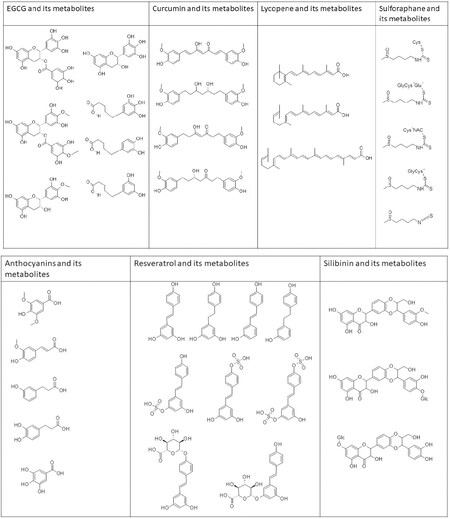
Figure 12. Chemical structures of phytochemicals and their metabolites. EGCG and metabolites - EGC, 4’-MeEGC; 4’,4’-diMeEGCG, 5-(3’, 4’-dihydroxyphenyl-γ-valerolactone (M6); 5-(3’, 4’, 5’- triihydroxyphenyl-γ-valerolactone (M4); 5-(3’, 5’- dihydroxyphenyl-γ-valerolactone (M6’). Curcumin and metabolites - tetrahydrocurcumin, hexahydrocurcumin, hexahydrocurcuminol. Lycopene and metabolites - apo-10’a-lycopenoic acid, acylclo-retinoic acid, all-trans-retinoic acid. Sulforaphane and metabolites - sulforapahane-GSH, sulforapahane cysteinylglycine. Anthocyanin and metabolites - syringic acid, ferulic acid 3,4-dihydroxyphenylpropionic acid, m-hydroxyphenylpropionic acid, gallic acid. Resveratrol and metabolites - trans-resveratrol (RSV), trans-resveratrol-3-O-sulfate, trans-resveratrol-4’-O-sulfate, trans-resveratrol-3,4’-O-disulfate, trans-resveratrol-3-O-glucuronide, trans-resveratrol-4’-O-glucuronide, dihydroresveratrol (DHR), 3,4’-O-dihydroxy-trans-stilbene, lunularin. Silibinin and metabolites - 20-O-β-D-Glu, 7-O-β-D-Glu, O-demethylated metabolite
In addition to well-known phase 1 and phase 2 mammalian enzymes that participate in drug metabolism, clearly, the overall metabolic profile of chemopreventive agents is influenced by the gut microbiome. Even further, however, health benefits mediated by phytochemicals may result from indirect effects such as influencing products produced by the gut microbiome itself. For example, recent studies have shown enrichment of short-chain fatty acids (SCFAs)-producing bacteria (e.g., Bacteroidetes and Parabacteroides) in mice treated with resveratrol[280]. SCFAs, microbial-derived metabolites, are reported to affect CNS functions in the microbiota-brain-axis[281].
Conversely, dietary chemopreventive agents may influence the actual composition, and therefore the metabolic capacity, of the gut microbiome. For example, in studies we have conducted with mice in which the diet was supplemented with whole grapes (unpublished), the overall organization of the gut microbiome was clearly altered [Figure 13]. This is yet another factor that needs to be considered in defining the overall action of chemopreventive agents.
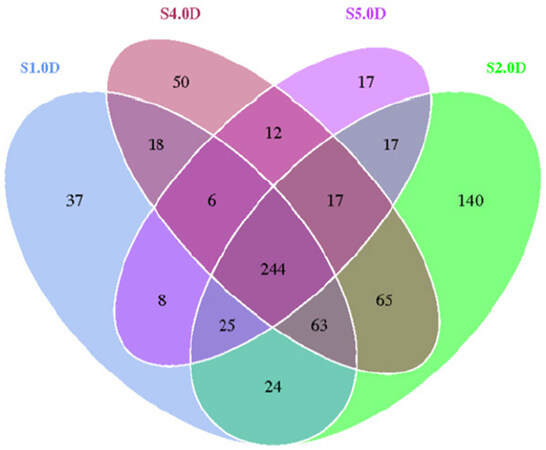
Figure 13. The figure shows a Venn diagram where each circle represents one group of microorganisms. The values in overlapping parts represent common Operational Taxonomic Units (OTUs) while others are OTUs specific to each group. Female HSD: ICR (CD-1®) mice at the age of 4 weeks were divided into four groups and given the following diets ad libitum for 20 weeks: S1.0D, standard synthetic diet, S2.0D; standard diet supplemented with 5% whole grape powder; S4.0D, high-fat (Western) diet;S5.0D, high-fat diet supplemented with 5% whole grape powder. The gut microbiota was analyzed using fecal samples. The data illustrate dietary grape supplementation influenced the composition of the gut microbiome. The overall organization of the gut microbiome is altered by grape and a higher number of microbiome groups can be observed in the grape fed diet groups
Sequelae
Over the past decades, there have been significant advancements in understanding cancers and in improving early detection, treatment, and prevention. As a result, the survival rate of cancer patients has increased. And, as the number of cancer survivors increases, the quality of life for these individuals must receive greater attention, given that adverse effects (sequelae) are frequently encountered during or after chemotherapy. One common sequela is cancer-related cognitive impairment (CRCI), or chemotherapy-induced cognitive impairments, colloquially called “chemobrain” or “chemofog”, a decline in cognitive function following cancer treatment. Central neurotoxicity can remain as long-term sequelae following the termination of treatment. For example, it has been reported that breast cancer and childhood cancer survivors may experience cognitive dysfunction[282]. Notably, some chemopreventive agents are at least touted to improve this condition[283,284].
Given the mode of action of chemopreventive agents, it seems logical that acute or chronic side-effects resulting from some forms of cancer chemotherapy could be ameliorated by concomitant administration. However, since chemotherapy is often designed to be overtly toxic, and therapeutic indices may be low, an effective chemopreventive agent could actually have an adverse effect on outcome by negating anti-cancer drug action. An appropriate balance is necessary, but some promising results have been described. For example, recent studies have demonstrated the neuroprotective potential of phytochemicals by reducing chemotherapy-induced CRCI in animal models. In addition, orally administered curcumin, at a dose of 100 mg/kg 1 h prior to cisplatin treatment, attenuated cisplatin-induced autophagy in the murine hippocampus accompanied by the activation of AMPK-JNK signaling[283]. Further, piceid, a resveratrol derivative, given orally at a dose of 50 mg/kg/d for 4 weeks, protected against doxorubicin-induced cognitive impairment concomitant with the up-regulation of Nrf2, inhibition of the NF-κB pathway, and reduction of apoptosis in the rat hippocampus[284]. It seems the ameliorative potential of chemoprevention is worthy of serious consideration.
In summary, over the past 50 years or so, many phytochemicals have been discovered and characterized that can delay, prevent, or reverse the process of carcinogenesis, and thereby modulate the incidence of the end stage cancer. Given the consequences of malignant metastatic disease, the promise of this therapeutic approach is evident yet remains largely untapped. As we continue to strive for primary intervention strategies that can be offered to all of those in need, additional potential benefits have become apparent, some of which are described herein and depicted in Figure 14. Not only does chemoprevention offer significant promise for disease prevention, but given the enormous economic burden imposed by contemporary biologic and drug therapies[285], the approach would be cost-effective.

Figure 14. Possible paradigm augmentation in cancer chemoprevention. As we realize shifts in the perspective of handling the dilemma of cancer, changing roles for chemopreventive agents can be envisioned. Emphasis is placed on sustaining “holistic health” and wellbeing
Declarations
AcknowledgmentsThe authors are grateful to the California Table Grape Commission for providing support for this work.
Authors’ contributionsParticipated in the conception and preparation of this review: Dave A, Parande F, Park EJ, Pezzuto JM
Availability of data and materialsNot applicable.
Financial support and sponsorshipThis work was supported in part by the California Table Grape Commission.
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2020.
REFERENCES
2. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 2018;68:394-424.
3. Chikara S, Nagaprashantha LD, Singhal J, Horne D, Awasthi S, Singhal SS. Oxidative stress and dietary phytochemicals: role in cancer chemoprevention and treatment. Cancer Lett 2018;413:122-34.
4. Madka V, Rao CV. Anti-inflammatory phytochemicals for chemoprevention of colon cancer. Curr Cancer Drug Targets 2013;13:542-57.
5. Pezzuto JM, Vang O. Natural products for cancer chemoprevention. Springer; 2020. pp. ix-xvi.
6. De Flora S, Ferguson LR. Overview of mechanisms of cancer chemopreventive agents. Mutat Res 2005;591:8-15.
7. Ball S, Arevalo M, Juarez E, Payne JD, Jones C. Breast cancer chemoprevention: an update on current practice and opportunities for primary care physicians. Prev Med 2019;129:105834.
8. Pezzuto JM, Vang O. Perspective: a positive cocktail effect of the bioactive components in the diet. Natural Products for Cancer Chemoprevention. Springer; 2020. p. 615.
10. Baena Ruiz R, Salinas Hernández P. Cancer chemoprevention by dietary phytochemicals: Epidemiological evidence. Maturitas 2016;94:13-9.
11. Howes MJ, Simmonds MS. The role of phytochemicals as micronutrients in health and disease. Curr Opin Clin Nutr Metab Care 2014;17:558-66.
12. Aunan JR, Cho WC, Søreide K. The biology of aging and cancer: a brief overview of shared and divergent molecular hallmarks. Aging Dis 2017;8:628-42.
13. Kotecha R, Takami A, Espinoza JL. Dietary phytochemicals and cancer chemoprevention: a review of the clinical evidence. Oncotarget 2016;7:52517-29.
14. Stevenson DE, Hurst RD. Polyphenolic phytochemicals--just antioxidants or much more? Cell Mol Life Sci 2007;64:2900-16.
15. Trela BC, Waterhouse AL. Resveratrol: isomeric molar absorptivities and stability. J Agric Food Chem 1996;44:1253-7.
16. Jeandet P, Douillet-Breuil AC, Bessis R, Debord S, Sbaghi M, Adrian M. Phytoalexins from the vitaceae: biosynthesis, phytoalexin gene expression in transgenic plants, antifungal activity, and metabolism. J Agric Food Chem 2002;50:2731-41.
17. Singh CK, Liu X, Ahmad N. Resveratrol, in its natural combination in whole grape, for health promotion and disease management. Ann N Y Acad Sci 2015;1348:150-60.
18. Maier-Salamon A, Böhmdorfer M, Thalhammer T, Szekeres T, Jaeger W. Hepatic glucuronidation of resveratrol: interspecies comparison of enzyme kinetic profiles in human, mouse, rat, and dog. Drug Metab Pharmacokinet 2011;26:364-73.
19. Soleas GJ, Diamandis EP, Goldberg DM. Resveratrol: a molecule whose time has come? And gone? Clin Biochem 1997;30:91-113.
20. Sato M, Maulik G, Bagchi D, Das DK. Myocardial protection by protykin, a novel extract of trans-resveratrol and emodin. Free Radic Res 2000;32:135-44.
21. Soleas GJ, Diamandis EP, Goldberg DM. Wine as a biological fluid: history, production, and role in disease prevention. J Clin Lab Anal 1997;11:287-313.
22. Nonomura S, Kanagawa H, Makimoto A. Chemical constituents of polygonaceous plants. I. studies on the components of Ko-J O-Kon. (Polygonum Cuspidatum Sieb. Et Zucc.). Yakugaku Zasshi 1963;83:988-90.
23. Chung MI, Teng CM, Cheng KL, Ko FN, Lin CN. An antiplatelet principle of veratrum formosanum. Planta Med 1992;58:274-6.
24. Crozier A, Jaganath IB, Clifford MN. Dietary phenolics: chemistry, bioavailability and effects on health. Nat Prod Rep 2009;26:1001-43.
25. Lançon A, Delmas D, Osman H, Thénot JP, Jannin B, Latruffe N. Human hepatic cell uptake of resveratrol: involvement of both passive diffusion and carrier-mediated process. Biochem Biophys Res Commun 2004;316:1132-7.
26. Wang LX, Heredia A, Song H, et al. Resveratrol glucuronides as the metabolites of resveratrol in humans: characterization, synthesis, and anti-HIV activity. J Pharm Sci 2004;93:2448-57.
27. Walle T, Hsieh F, DeLegge MH, Oatis JE, Walle UK. High absorption but very low bioavailability of oral resveratrol in humans. Drug Metab Dispos 2004;32:1377-82.
28. Boocock DJ, Patel KR, Faust GE, et al. Quantitation of trans-resveratrol and detection of its metabolites in human plasma and urine by high performance liquid chromatography. J Chromatogr B Analyt Technol Biomed Life Sci 2007;848:182-7.
29. Urpi-Sarda M, Zamora-Ros R, Lamuela-Raventos R, et al. HPLC-tandem mass spectrometric method to characterize resveratrol metabolism in humans. Clin Chem 2007;53:292-9.
30. Zamora-Ros R, Urpí-Sardà M, Lamuela-Raventós RM, et al. Diagnostic performance of urinary resveratrol metabolites as a biomarker of moderate wine consumption. Clin Chem 2006;52:1373-80.
31. Pattus F, Rothen C, Streit M, Zahler P. Structure, composition, enzymatic activities of human erythrocyte and sarcoplasmic reticulum membrane films. Biochim Biophys Acta 1981;647:29-39.
32. Yáñez M, Fraiz N, Cano E, Orallo F. Inhibitory effects of cis- and trans-resveratrol on noradrenaline and 5-hydroxytryptamine uptake and on monoamine oxidase activity. Biochem Biophys Res Commun 2006;344:688-95.
33. Wenzel E, Somoza V. Metabolism and bioavailability of trans-resveratrol. Mol Nutr Food Res 2005;49:472-81.
34. Renaud S, de Lorgeril M. Wine, alcohol, platelets, and the French paradox for coronary heart disease. Lancet 1992;339:1523-6.
36. Jang M, Cai L, Udeani GO, et al. Cancer chemopreventive activity of resveratrol, a natural product derived from grapes. Science 1997;275:218-20.
37. Pezzuto JM. Resveratrol: twenty years of growth, development and controversy. Biomol Ther (Seoul) 2019;27:1-14.
38. Gambini J, Inglés M, Olaso G, et al. Properties of resveratrol: in vitro and in vivo studies about metabolism, bioavailability, and biological effects in animal models and humans. Oxid Med Cell Longev 2015;2015:837042.
39. Pezzuto JM, Kondratyuk TP, Ogas T. Resveratrol derivatives: a patent review (2009 - 2012). Expert Opin Ther Pat 2013;23:1529-46.
40. Park EJ, Pezzuto JM. The pharmacology of resveratrol in animals and humans. Biochim Biophys Acta 2015;1852:1071-113.
41. Wadsworth TL, Koop DR. Effects of the wine polyphenolics quercetin and resveratrol on pro-inflammatory cytokine expression in RAW 264.7 macrophages. Biochem Pharmacol 1999;57:941-9.
42. Rotondo S, Rajtar G, Manarini S, et al. Effect of trans-resveratrol, a natural polyphenolic compound, on human polymorphonuclear leukocyte function. Br J Pharmacol 1998;123:1691-9.
43. Das S, Alagappan VK, Bagchi D, Sharma HS, Maulik N, Das DK. Coordinated induction of iNOS-VEGF-KDR-eNOS after resveratrol consumption: a potential mechanism for resveratrol preconditioning of the heart. Vascul Pharmacol 2005;42:281-9.
44. Imamura G, Bertelli AA, Bertelli A, Otani H, Maulik N, Das DK. Pharmacological preconditioning with resveratrol: an insight with iNOS knockout mice. Am J Physiol Heart Circ Physiol 2002;282:H1996-2003.
45. Kaneider NC, Mosheimer B, Reinisch N, Patsch JR, Wiedermann CJ. Inhibition of thrombin-induced signaling by resveratrol and quercetin: effects on adenosine nucleotide metabolism in endothelial cells and platelet-neutrophil interactions. Thromb Res 2004;114:185-94.
46. She QB, Bode AM, Ma WY, Chen NY, Dong Z. Resveratrol-induced activation of p53 and apoptosis is mediated by extracellular-signal-regulated protein kinases and p38 kinase. Cancer Res 2001;61:1604-10.
47. Ge H, Zhang JF, Guo BS, He B, Wang BY, Wang CQ. Resveratrol inhibits expression of EMMPRIN from macrophages. Yao Xue Xue Bao 2006;41:625-30.
48. Qin MZ, Gu QH, Tao J, Song XY, Gan GS, Luo ZB, Li BX. Ketamine effect on HMGB1 and TLR4 expression in rats with acute lung injury. Int J Clin Exp Pathol 2015;8:12943-8.
49. Jones BA, Beamer M, Ahmed S. Fractalkine/CX3CL1: a potential new target for inflammatory diseases. Mol Interv 2010;10:263-70.
50. Lee B, Moon SK. Resveratrol inhibits TNF-alpha-induced proliferation and matrix metalloproteinase expression in human vascular smooth muscle cells. J Nutr 2005;135:2767-73.
51. Takada Y, Bhardwaj A, Potdar P, Aggarwal BB. Nonsteroidal anti-inflammatory agents differ in their ability to suppress NF-kappaB activation, inhibition of expression of cyclooxygenase-2 and cyclin D1, and abrogation of tumor cell proliferation. Oncogene 2004;23:9247-58.
52. Szewczuk LM, Forti L, Stivala LA, Penning TM. Resveratrol is a peroxidase-mediated inactivator of COX-1 but not COX-2: a mechanistic approach to the design of COX-1 selective agents. J Biol Chem 2004;279:22727-37.
53. Subbaramaiah K, Chung WJ, Michaluart P, et al. Resveratrol inhibits cyclooxygenase-2 transcription and activity in phorbol ester-treated human mammary epithelial cells. J Biol Chem 1998;273:21875-82.
54. Martín AR, Villegas I, Sánchez-Hidalgo M, de la Lastra CA. The effects of resveratrol, a phytoalexin derived from red wines, on chronic inflammation induced in an experimentally induced colitis model. Br J Pharmacol 2006;147:873-85.
55. Lin HY, Lansing L, Merillon JM, et al. Integrin alphaVbeta3 contains a receptor site for resveratrol. FASEB J 2006;20:1742-4.
56. Nebert DW, Dalton TP, Okey AB, Gonzalez FJ. Role of aryl hydrocarbon receptor-mediated induction of the CYP1 enzymes in environmental toxicity and cancer. J Biol Chem 2004;279:23847-50.
57. Pozo-Guisado E, Merino JM, Mulero-Navarro S, Lorenzo-Benayas MJ, et al. Resveratrol-induced apoptosis in MCF-7 human breast cancer cells involves a caspase-independent mechanism with downregulation of Bcl-2 and NF-kappaB. Int J Cancer 2005;115:74-84.
58. Casper RF, Quesne M, Rogers IM, et al. Resveratrol has antagonist activity on the aryl hydrocarbon receptor: implications for prevention of dioxin toxicity. Mol Pharmacol 1999;56:784-90.
59. Hsieh TC, Wu JM. Differential effects on growth, cell cycle arrest, and induction of apoptosis by resveratrol in human prostate cancer cell lines. Exp Cell Res 1999;249:109-15.
60. Huang C, Ma WY, Goranson A, Dong Z. Resveratrol suppresses cell transformation and induces apoptosis through a p53-dependent pathway. Carcinogenesis 1999;20:237-42.
61. Moreno JJ. Resveratrol modulates arachidonic acid release, prostaglandin synthesis, and 3T6 fibroblast growth. J Pharmacol Exp Ther 2000;294:333-8.
62. Fontecave M, Lepoivre M, Elleingand E, Gerez C, Guittet O. Resveratrol, a remarkable inhibitor of ribonucleotide reductase. FEBS Lett 1998;421:277-9.
63. Peng L, Jiang D. Resveratrol eliminates cancer stem cells of osteosarcoma by STAT3 pathway inhibition. PLoS One 2018;13:e0205918.
64. Reto M, Figueira ME, Filipe HM, Almeida CM. Chemical composition of green tea (Camellia sinensis) infusions commercialized in Portugal. Plant Foods Hum Nutr 2007;62:139-44.
65. Chung FL, Schwartz J, Herzog CR, Yang YM. Tea and cancer prevention: studies in animals and humans. J Nutr 2003;133:3268S-74.
67. Sung H, Nah J, Chun S, Park H, Yang SE, Min WK. In vivo antioxidant effect of green tea. Eur J Clin Nutr 2000;54:527-9.
68. Ding S, Jiang J, Yu P, Zhang G, Liu X. Green tea polyphenol treatment attenuates atherosclerosis in high-fat diet-fed apolipoprotein E-knockout mice via alleviating dyslipidemia and up-regulating autophagy. PLoS One 2017;12:e0181666.
69. Reygaert WC. Green tea catechins: their use in treating and preventing infectious diseases. Biomed Res Int 2018;2018:9105261.
70. Sang S, Lambert JD, Ho CT, Yang CS. The chemistry and biotransformation of tea constituents. Pharmacol Res 2011;64:87-99.
71. Gan RY, Li HB, Sui ZQ, Corke H. Absorption, metabolism, anti-cancer effect and molecular targets of epigallocatechin gallate (EGCG): An updated review. Crit Rev Food Sci Nutr 2018;58:924-41.
72. Nakachi K, Suemasu K, Suga K, Takeo T, Imai K, Higashi Y. Influence of drinking green tea on breast cancer malignancy among Japanese patients. Jpn J Cancer Res 1998;89:254-61.
73. Uehara M, Sugiura H, Sakurai K. A trial of oolong tea in the management of recalcitrant atopic dermatitis. Arch Dermatol 2001;137:42-3.
74. Bushman JL. Green tea and cancer in humans: a review of the literature. Nutr Cancer 1998;31:151-9.
75. Rasheed A, Haider M. Antibacterial activity of Camellia sinensis extracts against dental caries. Arch Pharm Res 1998;21:348-52.
76. Guo YJ, Zhang B, Feng XS, Ren HX, Xu JR. Retraction Note: Human cathelicidin LL-37 enhance the antibiofilm effect of EGCG on Streptococcus mutans. BMC Oral Health 2017;17:102.
77. Kaya Z, Yayla M, Cinar I, et al. Epigallocatechin-3-gallate (EGCG) exert therapeutic effect on acute inflammatory otitis media in rats. Int J Pediatr Otorhinolaryngol 2019;124:106-10.
78. Zhu J, Jiang Y, Yang X, et al. Wnt/β-catenin pathway mediates (-)-Epigallocatechin-3-gallate (EGCG) inhibition of lung cancer stem cells. Biochem Biophys Res Commun 2017;482:15-21.
79. Toden S, Tran HM, Tovar-Camargo OA, Okugawa Y, Goel A. Epigallocatechin-3-gallate targets cancer stem-like cells and enhances 5-fluorouracil chemosensitivity in colorectal cancer. Oncotarget 2016;7:16158-71.
80. Jiang P, Xu C, Chen L, et al. EGCG inhibits CSC-like properties through targeting miR-485/CD44 axis in A549-cisplatin resistant cells. Mol Carcinog 2018;57:1835-44.
81. Sun Y, Liu WZ, Liu T, Feng X, Yang N, Zhou HF. Signaling pathway of MAPK/ERK in cell proliferation, differentiation, migration, senescence and apoptosis. J Recept Signal Transduct Res 2015;35:600-4.
82. Shirakami Y, Shimizu M. Possible mechanisms of green tea and its constituents against cancer. Molecules 2018;23:2284.
83. Bigelow RL, Cardelli JA. The green tea catechins, (-)-Epigallocatechin-3-gallate (EGCG) and (-)-Epicatechin-3-gallate (ECG), inhibit HGF/Met signaling in immortalized and tumorigenic breast epithelial cells. Oncogene 2006;25:1922-30.
84. Sah JF, Balasubramanian S, Eckert RL, Rorke EA. Epigallocatechin-3-gallate inhibits epidermal growth factor receptor signaling pathway. Evidence for direct inhibition of ERK1/2 and AKT kinases. J Biol Chem 2004;279:12755-62.
85. Khan N, Afaq F, Saleem M, Ahmad N, Mukhtar H. Targeting multiple signaling pathways by green tea polyphenol (-)-epigallocatechin-3-gallate. Cancer Res 2006;66:2500-5.
86. Jiang J, Mo ZC, Yin K, et al. Epigallocatechin-3-gallate prevents TNF-α-induced NF-κB activation thereby upregulating ABCA1 via the Nrf2/Keap1 pathway in macrophage foam cells. Int J Mol Med 2012;29:946-56.
87. Giakoustidis AE, Giakoustidis DE, Koliakou K, et al. Inhibition of intestinal ischemia/repurfusion induced apoptosis and necrosis via down-regulation of the NF-kB, c-Jun and caspace-3 expression by epigallocatechin-3-gallate administration. Free Radic Res 2008;42:180-8.
88. Sen T, Dutta A, Chatterjee A. Epigallocatechin-3-gallate (EGCG) downregulates gelatinase-B (MMP-9) by involvement of FAK/ERK/NFkappaB and AP-1 in the human breast cancer cell line MDA-MB-231. Anticancer Drugs 2010;21:632-44.
89. Kim HS, Kim MH, Jeong M, et al. EGCG blocks tumor promoter-induced MMP-9 expression via suppression of MAPK and AP-1 activation in human gastric AGS cells. Anticancer Res 2004;24:747-53.
90. Yan Z, Yong-Guang T, Fei-Jun L, Fa-Qing T, Min T, Ya C. Interference effect of epigallocatechin-3-gallate on targets of nuclear factor kappaB signal transduction pathways activated by EB virus encoded latent membrane protein 1. Int J Biochem Cell Biol 2004;36:1473-81.
91. Wu Q, Wu W, Fu B, Shi L, Wang X, Kuca K. JNK signaling in cancer cell survival. Med Res Rev 2019;39:2082-104.
92. Yamamoto T, Digumarthi H, Aranbayeva Z, et al. EGCG-targeted p57/KIP2 reduces tumorigenicity of oral carcinoma cells: role of c-Jun N-terminal kinase. Toxicol Appl Pharmacol 2007;224:318-25.
93. Cao G, Chen M, Song Q, et al. EGCG protects against UVB-induced apoptosis via oxidative stress and the JNK1/c-Jun pathway in ARPE19 cells. Mol Med Rep 2012;5:54-9.
94. Saeki K, Kobayashi N, Inazawa Y, et al. Oxidation-triggered c-Jun N-terminal kinase (JNK) and p38 mitogen-activated protein (MAP) kinase pathways for apoptosis in human leukaemic cells stimulated by epigallocatechin-3-gallate (EGCG): a distinct pathway from those of chemically induced and receptor-mediated apoptosis. Biochem J 2002;368:705-20.
95. Cuadrado A, Nebreda AR. Mechanisms and functions of p38 MAPK signalling. Biochemical Journal 2010;429:403-17.
96. Cerezo-Guisado MI, Zur R, Lorenzo MJ, et al. Implication of Akt, ERK1/2 and alternative p38MAPK signalling pathways in human colon cancer cell apoptosis induced by green tea EGCG. Food Chem Toxicol 2015;84:125-32.
97. Kim SJ, Jeong HJ, Lee KM, et al. Epigallocatechin-3-gallate suppresses NF-kappaB activation and phosphorylation of p38 MAPK and JNK in human astrocytoma U373MG cells. J Nutr Biochem 2007;18:587-96.
98. Kang YH, Pezzuto JM. Induction of quinone reductase as a primary screen for natural product anticarcinogens. Methods Enzymol 2004;382:380-414.
99. Fahey JW, Talalay P. Antioxidant functions of sulforaphane: a potent inducer of Phase II detoxication enzymes. Food Chem Toxicol 1999;37:973-9.
100. Lenzi M, Fimognari C, Hrelia P. Sulforaphane as a promising molecule for fighting cancer. Cancer Treat Res 2014;159:207-23.
101. Kuljarachanan T, Fu N, Chiewchan N, Devahastin S, Chen XD. Evolution of important glucosinolates in three common Brassica vegetables during their processing into vegetable powder and in vitro gastric digestion. Food Funct 2020;11:211-20.
102. Matusheski NV, Swarup R, Juvik JA, Mithen R, Bennett M, Jeffery EH. Epithiospecifier protein from broccoli (Brassica oleracea L. ssp. italica) inhibits formation of the anticancer agent sulforaphane. J Agric Food Chem 2006;54:2069-76.
103. Matusheski NV, Juvik JA, Jeffery EH. Heating decreases epithiospecifier protein activity and increases sulforaphane formation in broccoli. Phytochemistry 2004;65:1273-81.
104. Gasper AV, Al-Janobi A, Smith JA, et al. Glutathione S-transferase M1 polymorphism and metabolism of sulforaphane from standard and high-glucosinolate broccoli. Am J Clin Nutr 2005;82:1283-91.
105. Egner PA, Chen JG, Wang JB, et al. Bioavailability of Sulforaphane from two broccoli sprout beverages: results of a short-term, cross-over clinical trial in Qidong, China. Cancer Prev Res (Phila) 2011;4:384-95.
106. Houghton CA, Fassett RG, Coombes JS. Sulforaphane: translational research from laboratory bench to clinic. Nutr Rev 2013;71:709-26.
107. Yagishita Y, Fahey JW, Dinkova-Kostova AT, Kensler TW. Broccoli or sulforaphane: is it the source or dose that matters? Molecules 2019;24:3593.
108. Huang C, Wu J, Chen D, Jin J, Wu Y, Chen Z. Effects of sulforaphane in the central nervous system. Eur J Pharmacol 2019;853:153-68.
109. Shawky NM, Shehatou GSG, Suddek GM, Gameil NM. Comparison of the effects of sulforaphane and pioglitazone on insulin resistance and associated dyslipidemia, hepatosteatosis, and endothelial dysfunction in fructose-fed rats. Environ Toxicol Pharmacol 2019;66:43-54.
110. Bahadoran Z, Tohidi M, Nazeri P, Mehran M, Azizi F, Mirmiran P. Effect of broccoli sprouts on insulin resistance in type 2 diabetic patients: a randomized double-blind clinical trial. Int J Food Sci Nutr 2012;63:767-71.
111. Axelsson AS, Tubbs E, Mecham B, et al. Sulforaphane reduces hepatic glucose production and improves glucose control in patients with type 2 diabetes. Sci Transl Med 2017;9:eaah4477.
112. Murashima M, Watanabe S, Zhuo XG, Uehara M, Kurashige A. Phase 1 study of multiple biomarkers for metabolism and oxidative stress after one-week intake of broccoli sprouts. Biofactors 2004;22:271-5.
113. Mazarakis N, Snibson K, Licciardi PV, Karagiannis TC. The potential use of l-sulforaphane for the treatment of chronic inflammatory diseases: a review of the clinical evidence. Clin Nutr 2020;39:664-75.
114. Liu H, Talalay P. Relevance of anti-inflammatory and antioxidant activities of exemestane and synergism with sulforaphane for disease prevention. Proc Natl Acad Sci U S A 2013;110:19065-70.
115. Clarke JD, Hsu A, Yu Z, Dashwood RH, Ho E. Differential effects of sulforaphane on histone deacetylases, cell cycle arrest and apoptosis in normal prostate cells versus hyperplastic and cancerous prostate cells. Mol Nutr Food Res 2011;55:999-1009.
116. Cornblatt BS, Ye L, Dinkova-Kostova AT, et al. Preclinical and clinical evaluation of sulforaphane for chemoprevention in the breast. Carcinogenesis 2007;28:1485-90.
117. Carpenter CL, Yu MC, London SJ. Dietary isothiocyanates, glutathione S-transferase M1 (GSTM1), and lung cancer risk in African Americans and Caucasians from Los Angeles County, California. Nutr Cancer 2009;61:492-9.
118. Wu QJ, Xie L, Zheng W, et al. Cruciferous vegetables consumption and the risk of female lung cancer: a prospective study and a meta-analysis. Ann Oncol 2013;24:1918-24.
119. Li QQ, Xie YK, Wu Y, et al. Sulforaphane inhibits cancer stem-like cell properties and cisplatin resistance through miR-214-mediated downregulation of c-MYC in non-small cell lung cancer. Oncotarget 2017;8:12067-80.
120. Juge N, Mithen RF, Traka M. Molecular basis for chemoprevention by sulforaphane: a comprehensive review. Cell Mol Life Sci 2007;64:1105-27.
121. Jin W, Wang H, Ji Y, et al. Genetic ablation of Nrf2 enhances susceptibility to acute lung injury after traumatic brain injury in mice. Exp Biol Med (Maywood) 2009;234:181-9.
122. Kong X, Thimmulappa R, Kombairaju P, Biswal S. NADPH oxidase-dependent reactive oxygen species mediate amplified TLR4 signaling and sepsis-induced mortality in Nrf2-deficient mice. J Immunol 2010;185:569-77.
123. Dinkova-Kostova AT, Fahey JW, Kostov RV, Kensler TW. KEAP1 and done? Targeting the NRF2 Pathway with sulforaphane. Trends Food Sci Technol 2017;69:257-69.
124. Thimmulappa RK, Mai KH, Srisuma S, Kensler TW, Yamamoto M, Biswal S. Identification of Nrf2-regulated genes induced by the chemopreventive agent sulforaphane by oligonucleotide microarray. Cancer Res 2002;62:5196-203.
125. Alao JP. The regulation of cyclin D1 degradation: roles in cancer development and the potential for therapeutic invention. Mol Cancer 2007;6:24.
126. Żuryń A, Litwiniec A, Safiejko-Mroczka B, et al. The effect of sulforaphane on the cell cycle, apoptosis and expression of cyclin D1 and p21 in the A549 non-small cell lung cancer cell line. Int J Oncol 2016;48:2521-33.
127. Wang L, Liu D, Ahmed T, Chung FL, Conaway C, Chiao JW. Targeting cell cycle machinery as a molecular mechanism of sulforaphane in prostate cancer prevention. Int J Oncol 2004;24:187-92.
128. Hu R, Khor TO, Shen G, et al. Cancer chemoprevention of intestinal polyposis in ApcMin/+ mice by sulforaphane, a natural product derived from cruciferous vegetable. Carcinogenesis 2006;27:2038-46.
129. Ho E, Clarke JD, Dashwood RH. Dietary sulforaphane, a histone deacetylase inhibitor for cancer prevention. J Nutr 2009;139:2393-6.
130. Myzak MC, Hardin K, Wang R, Dashwood RH, Ho E. Sulforaphane inhibits histone deacetylase activity in BPH-1, LnCaP and PC-3 prostate epithelial cells. Carcinogenesis 2006;27:811-9.
131. Jiang LL, Zhou SJ, Zhang XM, Chen HQ, Liu W. Sulforaphane suppresses in vitro and in vivo lung tumorigenesis through downregulation of HDAC activity. Biomed Pharmacother 2016;78:74-80.
132. Myzak MC, Tong P, Dashwood WM, Dashwood RH, Ho E. Sulforaphane retards the growth of human PC-3 xenografts and inhibits HDAC activity in human subjects. Exp Biol Med (Maywood) 2007;232:227-34.
133. Juengel E, Erb HHH, Haferkamp A, Rutz J, Chun FK, Blaheta RA. Relevance of the natural HDAC inhibitor sulforaphane as a chemopreventive agent in urologic tumors. Cancer Lett 2018;435:121-6.
134. Alumkal JJ, Slottke R, Schwartzman J, et al. A phase II study of sulforaphane-rich broccoli sprout extracts in men with recurrent prostate cancer. Invest New Drugs 2015;33:480-9.
135. Cipolla BG, Mandron E, Lefort JM, et al. Effect of Sulforaphane in Men with Biochemical Recurrence after Radical Prostatectomy. Cancer Prev Res (Phila) 2015;8:712-9.
136. Bunea A, Rugină D, Sconţa Z, et al. Anthocyanin determination in blueberry extracts from various cultivars and their antiproliferative and apoptotic properties in B16-F10 metastatic murine melanoma cells. Phytochemistry 2013;95:436-44.
137. Konczak I, Zhang W. Anthocyanins-more than nature’s colours. J Biomed Biotechnol 2004;2004:239-40.
138. Kong JM, Chia LS, Goh NK, Chia TF, Brouillard R. Analysis and biological activities of anthocyanins. Phytochemistry 2003;64:923-33.
139. Hou DX, Kai K, Li JJ, et al. Anthocyanidins inhibit activator protein 1 activity and cell transformation: structure-activity relationship and molecular mechanisms. Carcinogenesis 2004;25:29-36.
140. Xu M, Bower KA, Wang S, et al. Cyanidin-3-glucoside inhibits ethanol-induced invasion of breast cancer cells overexpressing ErbB2. Mol Cancer 2010;9:285.
141. Dong Z, Birrer MJ, Watts RG, Matrisian LM, Colburn NH. Blocking of tumor promoter-induced AP-1 activity inhibits induced transformation in JB6 mouse epidermal cells. Proc Natl Acad Sci U S A 1994;91:609-13.
142. Huang C, Ma WY, Young MR, Colburn N, Dong Z. Shortage of mitogen-activated protein kinase is responsible for resistance to AP-1 transactivation and transformation in mouse JB6 cells. Proc Natl Acad Sci U S A 1998;95:156-61.
143. Chinery R, Beauchamp RD, Shyr Y, Kirkland SC, Coffey RJ, Morrow JD. Antioxidants reduce cyclooxygenase-2 expression, prostaglandin production, and proliferation in colorectal cancer cells. Cancer Res 1998;58:2323-7.
144. Hou DX, Fujii M, Terahara N, Yoshimoto M. Molecular Mechanisms behind the chemopreventive effects of anthocyanidins. J Biomed Biotechnol 2004;2004:321-5.
145. Hou DX, Ose T, Lin S, et al. Anthocyanidins induce apoptosis in human promyelocytic leukemia cells: structure-activity relationship and mechanisms involved. Int J Oncol 2003;23:705-12.
146. Li D, Zhang Y, Liu Y, Sun R, Xia M. Purified anthocyanin supplementation reduces dyslipidemia, enhances antioxidant capacity, and prevents insulin resistance in diabetic patients. J Nutr 2015;145:742-8.
147. Shih PH, Yeh CT, Yen GC. Anthocyanins induce the activation of phase II enzymes through the antioxidant response element pathway against oxidative stress-induced apoptosis. J Agric Food Chem 2007;55:9427-35.
148. Yi L, Chen CY, Jin X, et al. Structural requirements of anthocyanins in relation to inhibition of endothelial injury induced by oxidized low-density lipoprotein and correlation with radical scavenging activity. FEBS Lett 2010;584:583-90.
149. Esposito D, Chen A, Grace MH, Komarnytsky S, Lila MA. Inhibitory effects of wild blueberry anthocyanins and other flavonoids on biomarkers of acute and chronic inflammation in vitro. J Agric Food Chem 2014;62:7022-8.
150. Peiffer DS, Zimmerman NP, Wang LS, et al. Chemoprevention of esophageal cancer with black raspberries, their component anthocyanins, and a major anthocyanin metabolite, protocatechuic acid. Cancer Prev Res (Phila) 2014;7:574-84.
151. Charepalli V, Reddivari L, Radhakrishnan S, Vadde R, Agarwal R, Vanamala JK. Anthocyanin-containing purple-fleshed potatoes suppress colon tumorigenesis via elimination of colon cancer stem cells. J Nutr Biochem 2015;26:1641-9.
152. Fimognari C, Berti F, Nüsse M, Cantelli-Forti G, Hrelia P. Induction of apoptosis in two human leukemia cell lines as well as differentiation in human promyelocytic cells by cyanidin-3-O-beta-glucopyranoside. Biochem Pharmacol 2004;67:2047-56.
153. Chang YC, Huang HP, Hsu JD, Yang SF, Wang CJ. Hibiscus anthocyanins rich extract-induced apoptotic cell death in human promyelocytic leukemia cells. Toxicol Appl Pharmacol 2005;205:201-12.
154. Oak MH, Bedoui JE, Madeira SV, Chalupsky K, Schini-Kerth VB. Delphinidin and cyanidin inhibit PDGF(AB)-induced VEGF release in vascular smooth muscle cells by preventing activation of p38 MAPK and JNK. Br J Pharmacol 2006;149:283-90.
155. Breinholt VM, Offord EA, Brouwer C, Nielsen SE, Brøsen K, Friedberg T. In vitro investigation of cytochrome P450-mediated metabolism of dietary flavonoids. Food Chem Toxicol 2002;40:609-16.
156. Mazza G, Kay CD, Cottrell T, Holub BJ. Absorption of anthocyanins from blueberries and serum antioxidant status in human subjects. J Agric Food Chem 2002;50:7731-7.
157. Kay CD, Mazza GJ, Holub BJ. Anthocyanins exist in the circulation primarily as metabolites in adult men. J Nutr 2005;135:2582-8.
158. Shimoi K, Okada H, Furugori M, et al. Intestinal absorption of luteolin and luteolin 7-O-beta-glucoside in rats and humans. FEBS Lett 1998;438:220-4.
159. Donovan JL, Crespy V, Manach C, et al. Catechin is metabolized by both the small intestine and liver of rats. J Nutr 2001;131:1753-7.
160. Spencer JP, Chowrimootoo G, Choudhury R, Debnam ES, Srai SK, Rice-Evans C. The small intestine can both absorb and glucuronidate luminal flavonoids. FEBS Lett 1999;458:224-30.
161. Oliveira EJ, Watson DG, Grant MH. Metabolism of quercetin and kaempferol by rat hepatocytes and the identification of flavonoid glycosides in human plasma. Xenobiotica 2002;32:279-87.
162. Kuhnle G, Spencer JP, Schroeter H, et al. Epicatechin and catechin are O-methylated and glucuronidated in the small intestine. Biochem Biophys Res Commun 2000;277:507-12.
163. Mojarrabi B, Mackenzie PI. Characterization of two UDP glucuronosyltransferases that are predominantly expressed in human colon. Biochem Biophys Res Commun 1998;247:704-9.
164. Strassburg CP, Nguyen N, Manns MP, Tukey RH. Polymorphic expression of the UDP-glucuronosyltransferase UGT1A gene locus in human gastric epithelium. Mol Pharmacol 1998;54:647-54.
165. Cheng Z, Radominska-Pandya A, Tephly TR. Studies on the substrate specificity of human intestinal UDP- lucuronosyltransferases 1A8 and 1A10. Drug Metab Dispos 1999;27:1165-70.
166. Crespy V, Morand C, Manach C, Besson C, Demigne C, Remesy C. Part of quercetin absorbed in the small intestine is conjugated and further secreted in the intestinal lumen. Am J Physiol 1999;277:G120-6.
167. Piskula MK, Terao J. Accumulation of (-)-epicatechin metabolites in rat plasma after oral administration and distribution of conjugation enzymes in rat tissues. J Nutr 1998;128:1172-8.
168. Manach C, Morand C, Crespy V, et al. Quercetin is recovered in human plasma as conjugated derivatives which retain antioxidant properties. FEBS Lett 1998;426:331-6.
169. Coughtrie MW, Sharp S, Maxwell K, Innes NP. Biology and function of the reversible sulfation pathway catalysed by human sulfotransferases and sulfatases. Chem Biol Interact 1998;109:3-27.
170. Del Rio D, Borges G, Crozier A. Berry flavonoids and phenolics: bioavailability and evidence of protective effects. Br J Nutr 2010;104 Suppl 3:S67-90.
171. Grynkiewicz G, Ślifirski P. Curcumin and curcuminoids in quest for medicinal status. Acta Biochimica Polonica 2012;59:201-12.
172. Kocaadam B, Şanlier N. Curcumin, an active component of turmeric (Curcuma longa), and its effects on health. Crit Rev Food Sci Nutr 2017;57:2889-95.
173. Priyadarsini KI. The chemistry of curcumin: from extraction to therapeutic agent. Molecules 2014;19:20091-112.
174. Nelson KM, Dahlin JL, Bisson J, Graham J, Pauli GF, Walters MA. The essential medicinal chemistry of curcumin. J Med Chem 2017;60:1620-37.
175. Priyadarsini KI. Chemical and structural features influencing the biological activity of curcumin. Curr Pharm Des 2013;19:2093-100.
176. Yang KY, Lin LC, Tseng TY, Wang SC, Tsai TH. Oral bioavailability of curcumin in rat and the herbal analysis from Curcuma longa by LC-MS/MS. J Chromatogr B Analyt Technol Biomed Life Sci 2007;853:183-9.
177. Lao CD, Ruffin MT, Normolle D, et al. Dose escalation of a curcuminoid formulation. BMC Complement Altern Med 2006;6:10.
178. Vareed SK, Kakarala M, Ruffin MT, et al. Pharmacokinetics of curcumin conjugate metabolites in healthy human subjects. Cancer Epidemiol Biomarkers Prev 2008;17:1411-7.
179. Ireson CR, Jones DJ, Orr S, et al. Metabolism of the cancer chemopreventive agent curcumin in human and rat intestine. Cancer Epidemiol Biomarkers Prev 2002;11:105-11.
180. Panahi Y, Fazlolahzadeh O, Atkin SL, et al. Evidence of curcumin and curcumin analogue effects in skin diseases: a narrative review. J Cell Physiol 2019;234:1165-78.
181. Krausz AE, Adler BL, Cabral V, et al. Curcumin-encapsulated nanoparticles as innovative antimicrobial and wound healing agent. Nanomedicine 2015;11:195-206.
182. Kang D, Li B, Luo L, et al. Curcumin shows excellent therapeutic effect on psoriasis in mouse model. Biochimie 2016;123:73-80.
183. Islam T, Koboziev I, Scoggin S, Ramalingam L, Moustaid-Moussa N. Protective effects of curcumin in high fat diet (HFD)-induced obesity include anti-inflammatory effects in adipose tissue and changes in gut microbiome (P06-075-19). Curr Dev Nutr 2019;3:nzz031-P06.
184. Henrotin Y, Gharbi M, Dierckxsens Y, et al. Decrease of a specific biomarker of collagen degradation in osteoarthritis, Coll2-1, by treatment with highly bioavailable curcumin during an exploratory clinical trial. BMC Complement Altern Med 2014;14:159.
185. Chandran B, Goel A. A randomized, pilot study to assess the efficacy and safety of curcumin in patients with active rheumatoid arthritis. Phytother Res 2012;26:1719-25.
186. Wu S, Xiao D. Effect of curcumin on nasal symptoms and airflow in patients with perennial allergic rhinitis. Ann Allergy Asthma Immunol 2016;117:697-702.e1.
187. Ledda A, Belcaro G, Dugall M, et al. Meriva®, a lecithinized curcumin delivery system, in the control of benign prostatic hyperplasia: a pilot, product evaluation registry study. Panminerva Med 2012;54:17-22.
188. Ide H, Tokiwa S, Sakamaki K, et al. Combined inhibitory effects of soy isoflavones and curcumin on the production of prostate-specific antigen. Prostate 2010;70:1127-33.
189. Bayet-Robert M, Kwiatkowski F, Leheurteur M, et al. Phase I dose escalation trial of docetaxel plus curcumin in patients with advanced and metastatic breast cancer. Cancer Biol Ther 2010;9:8-14.
190. Basu P, Dutta S, Begum R, et al. Clearance of cervical human papillomavirus infection by topical application of curcumin and curcumin containing polyherbal cream: a phase II randomized controlled study. Asian Pac J Cancer Prev 2013;14:5753-9.
191. Kanai M, Yoshimura K, Asada M, et al. A phase I/II study of gemcitabine-based chemotherapy plus curcumin for patients with gemcitabine-resistant pancreatic cancer. Cancer Chemother Pharmacol 2011;68:157-64.
192. Dhillon N, Aggarwal BB, Newman RA, et al. Phase II trial of curcumin in patients with advanced pancreatic cancer. Clin Cancer Res 2008;14:4491-9.
193. Saberi-Karimian M, Katsiki N, Caraglia M, et al. Vascular endothelial growth factor: an important molecular target of curcumin. Crit Rev Food Sci Nutr 2019;59:299-312.
194. Pan Z, Zhuang J, Ji C, Cai Z, Liao W, Huang Z. Curcumin inhibits hepatocellular carcinoma growth by targeting VEGF expression. Oncol Lett 2018;15:4821-6.
195. Da W, Zhang J, Zhang R, Zhu J. Curcumin inhibits the lymphangiogenesis of gastric cancer cells by inhibiton of HMGB1/VEGF-D signaling. Int J Immunopathol Pharmacol 2019;33:2058738419861600.
196. Srivastava RK, Chen Q, Siddiqui I, Sarva K, Shankar S. Linkage of curcumin-induced cell cycle arrest and apoptosis by cyclin-dependent kinase inhibitor p21(/WAF1/CIP1). Cell Cycle 2007;6:2953-61.
197. Lee DS, Lee MK, Kim JH. Curcumin induces cell cycle arrest and apoptosis in human osteosarcoma (HOS) cells. Anticancer Res 2009;29:5039-44.
198. Shishodia S, Amin HM, Lai R, Aggarwal BB. Curcumin (diferuloylmethane) inhibits constitutive NF-kappaB activation, induces G1/S arrest, suppresses proliferation, and induces apoptosis in mantle cell lymphoma. Biochem Pharmacol 2005;70:700-13.
199. Baharuddin P, Satar N, Fakiruddin KS, et al. Curcumin improves the efficacy of cisplatin by targeting cancer stem-like cells through p21 and cyclin D1-mediated tumour cell inhibition in non-small cell lung cancer cell lines. Oncol Rep 2016;35:13-25.
200. Aoki H, Takada Y, Kondo S, Sawaya R, Aggarwal BB, Kondo Y. Evidence that curcumin suppresses the growth of malignant gliomas in vitro and in vivo through induction of autophagy: role of Akt and extracellular signal-regulated kinase signaling pathways. Mol Pharmacol 2007;72:29-39.
201. Hussain AR, Al-Rasheed M, Manogaran PS, et al. Curcumin induces apoptosis via inhibition of PI3’-kinase/AKT pathway in acute T cell leukemias. Apoptosis 2006;11:245-54.
202. Bhatia N, Zhao J, Wolf DM, Agarwal R. Inhibition of human carcinoma cell growth and DNA synthesis by silibinin, an active constituent of milk thistle: comparison with silymarin. Cancer Lett 1999;147:77-84.
203. Gu W, Chen X, Pan X, Chan ASC, Yang TK. First enantioselective syntheses of (2R, 3R)-and (2S, 3S)-3-(4-hydroxy-3-methoxyphenyl)-2-hydroxymethyl-1, 4-benzodioxan-6-carbaldehyde. Tetrahedron Asymmetry 2000;11:2801-7.
204. Davis-Searles PR, Nakanishi Y, Kim NC, et al. Milk thistle and prostate cancer: differential effects of pure flavonolignans from Silybum marianum on antiproliferative end points in human prostate carcinoma cells. Cancer Res 2005;65:4448-57.
205. Luper S. A review of plants used in the treatment of liver disease: part 1. Altern Med Rev 1998;3:410-21.
206. Lettéron P, Labbe G, Degott C, et al. Mechanism for the protective effects of silymarin against carbon tetrachloride-induced lipid peroxidation and hepatotoxicity in mice. Evidence that silymarin acts both as an inhibitor of metabolic activation and as a chain-breaking antioxidant. Biochem Pharmacol 1990;39:2027-34.
207. Mereish KA, Bunner DL, Ragland DR, Creasia DA. Protection against microcystin-LR-induced hepatotoxicity by Silymarin: biochemistry, histopathology, and lethality. Pharm Res 1991;8:273-7.
208. Mourelle M, Muriel P, Favari L, Franco T. Prevention of CCL4-induced liver cirrhosis by silymarin. Fundam Clin Pharmacol 1989;3:183-91.
209. Valenzuela A, Guerra R, Videla LA. Antioxidant properties of the flavonoids silybin and (+)-cyanidanol-3: comparison with butylated hydroxyanisole and butylated hydroxytoluene. Planta Med 1986:438-40.
210. Deep G, Agarwal R. Antimetastatic efficacy of silibinin: molecular mechanisms and therapeutic potential against cancer. Cancer Metastasis Rev 2010;29:447-63.
211. Kumar R, Deep G, Agarwal R. An overview of ultraviolet b radiation-induced skin cancer chemoprevention by silibinin. Curr Pharmacol Rep 2015;1:206-15.
212. Ramasamy K, Agarwal R. Multitargeted therapy of cancer by silymarin. Cancer Lett 2008;269:352-62.
213. Redondo-Blanco S, Fernández J, Gutiérrez-Del-Río I, Villar CJ, Lombó F. New insights toward colorectal cancer chemotherapy using natural bioactive compounds. Front Pharmacol 2017;8:109.
214. Wen Z, Dumas TE, Schrieber SJ, Hawke RL, Fried MW, Smith PC. Pharmacokinetics and metabolic profile of free, conjugated, and total silymarin flavonolignans in human plasma after oral administration of milk thistle extract. Drug Metab Dispos 2008;36:65-72.
215. Flora K, Hahn M, Rosen H, Benner K. Milk thistle (Silybum marianum) for the therapy of liver disease. Am J Gastroenterol 1998;93:139-43.
216. Saller R, Meier R, Brignoli R. The use of silymarin in the treatment of liver diseases. Drugs 2001;61:2035-63.
217. Saller R, Melzer J, Reichling J, Brignoli R, Meier R. An updated systematic review of the pharmacology of silymarin. Forsch Komplementmed 2007;14:70-80.
218. Hawke RL, Schrieber SJ, Soule TA, et al. Silymarin ascending multiple oral dosing phase I study in noncirrhotic patients with chronic hepatitis C. J Clin Pharmacol 2010;50:434-49.
219. Kidd PM. Bioavailability and activity of phytosome complexes from botanical polyphenols: the silymarin, curcumin, green tea, and grape seed extracts. Altern Med Rev 2009;14:226-46.
220. Wang Y, Zhang D, Liu Z, et al. In vitro and in vivo evaluation of silybin nanosuspensions for oral and intravenous delivery. Nanotechnology 2010;21:155104.
221. Pietta P, Simonetti P, Gardana C, Brusamolino A, Morazzoni P, Bombardelli E. Relationship between rate and extent of catechin absorption and plasma antioxidant status. Biochem Mol Biol Int 1998;46:895-903.
222. Morazzoni P, Montalbetti A, Malandrino S, Pifferi G. Comparative pharmacokinetics of silipide and silymarin in rats. Eur J Drug Metab Pharmacokinet 1993;18:289-97.
223. Flaig TW, Glodé M, Gustafson D, et al. A study of high-dose oral silybin-phytosome followed by prostatectomy in patients with localized prostate cancer. Prostate 2010;70:848-55.
224. Flaig TW, Gustafson DL, Su LJ, et al. A phase I and pharmacokinetic study of silybin-phytosome in prostate cancer patients. Invest New Drugs 2007;25:139-46.
225. Hoh C, Boocock D, Marczylo T, et al. Pilot study of oral silibinin, a putative chemopreventive agent, in colorectal cancer patients: silibinin levels in plasma, colorectum, and liver and their pharmacodynamic consequences. Clin Cancer Res 2006;12:2944-50.
226. Chu SC, Chiou HL, Chen PN, Yang SF, Hsieh YS. Silibinin inhibits the invasion of human lung cancer cells via decreased productions of urokinase-plasminogen activator and matrix metalloproteinase-2. Mol Carcinog 2004;40:143-9.
227. Mateen S, Tyagi A, Agarwal C, Singh RP, Agarwal R. Silibinin inhibits human nonsmall cell lung cancer cell growth through cell-cycle arrest by modulating expression and function of key cell-cycle regulators. Mol Carcinog 2010;49:247-58.
228. Dagne A, Melkamu T, Schutten MM, Qian X, Upadhyaya P, Luo X, Kassie F. Enhanced inhibition of lung adenocarcinoma by combinatorial treatment with indole-3-carbinol and silibinin in A/J mice. Carcinogenesis 2011;32:561-7.
229. Corominas-Faja B, Oliveras-Ferraros C, Cuyàs E, et al. Stem cell-like ALDH(bright) cellular states in EGFR-mutant non-small cell lung cancer: a novel mechanism of acquired resistance to erlotinib targetable with the natural polyphenol silibinin. Cell Cycle 2013;12:3390-404.
230. Zi X, Agarwal R. Silibinin decreases prostate-specific antigen with cell growth inhibition via G1 arrest, leading to differentiation of prostate carcinoma cells: implications for prostate cancer intervention. Proc Natl Acad Sci U S A 1999;96:7490-5.
231. Zi X, Zhang J, Agarwal R, Pollak M. Silibinin up-regulates insulin-like growth factor-binding protein 3 expression and inhibits proliferation of androgen-independent prostate cancer cells. Cancer Res 2000;60:5617-20.
232. Mokhtari MJ, Motamed N, Shokrgozar MA. Evaluation of silibinin on the viability, migration and adhesion of the human prostate adenocarcinoma (PC-3) cell line. Cell Biol Int 2008;32:888-92.
233. Wu KJ, Zeng J, Zhu GD, et al. Silibinin inhibits prostate cancer invasion, motility and migration by suppressing vimentin and MMP-2 expression. Acta Pharmacol Sin 2009;30:1162-8.
234. Wu K, Zeng J, Li L, et al. Silibinin reverses epithelial-to-mesenchymal transition in metastatic prostate cancer cells by targeting transcription factors. Oncol Rep 2010;23:1545-52.
235. Kaur M, Velmurugan B, Tyagi A, et al. Silibinin suppresses growth and induces apoptotic death of human colorectal carcinoma LoVo cells in culture and tumor xenograft. Mol Cancer Ther 2009;8:2366-74.
236. Singh RP, Gu M, Agarwal R. Silibinin inhibits colorectal cancer growth by inhibiting tumor cell proliferation and angiogenesis. Cancer Res 2008;68:2043-50.
237. Kauntz H, Bousserouel S, Gossé F, Raul F. The flavonolignan silibinin potentiates TRAIL-induced apoptosis in human colon adenocarcinoma and in derived TRAIL-resistant metastatic cells. Apoptosis 2012;17:797-809.
238. van Breemen RB, Pajkovic N. Multitargeted therapy of cancer by lycopene. Cancer Lett 2008;269:339-51.
239. Clinton SK. Lycopene: chemistry, biology, and implications for human health and disease. Nutr Rev 1998;56:35-51.
240. Story EN, Kopec RE, Schwartz SJ, Harris GK. An update on the health effects of tomato lycopene. Annu Rev Food Sci Technol 2010;1:189-210.
241. Rissanen TH, Voutilainen S, Nyyssönen K, Salonen R, Kaplan GA, Salonen JT. Serum lycopene concentrations and carotid atherosclerosis: the kuopio ischaemic heart disease risk factor study. Am J Clin Nutr 2003;77:133-8.
242. Di Mascio P, Kaiser S, Sies H. Lycopene as the most efficient biological carotenoid singlet oxygen quencher. Arch Biochem Biophys 1989;274:532-8.
243. Muzandu K, Ishizuka M, Sakamoto KQ, et al. Effect of lycopene and beta-carotene on peroxynitrite-mediated cellular modifications. Toxicol Appl Pharmacol 2006;215:330-40.
244. Muzandu K, El Bohi K, Shaban Z, Ishizuka M, Kazusaka A, Fujita S. Lycopene and beta-carotene ameliorate catechol estrogen-mediated DNA damage. Jpn J Vet Res 2005;52:173-84.
245. Liu A, Pajkovic N, Pang Y, et al. Absorption and subcellular localization of lycopene in human prostate cancer cells. Mol Cancer Ther 2006;5:2879-85.
246. Park YO, Hwang ES, Moon TW. The effect of lycopene on cell growth and oxidative DNA damage of Hep3B human hepatoma cells. Biofactors 2005;23:129-39.
247. Hantz HL, Young LF, Martin KR. Physiologically attainable concentrations of lycopene induce mitochondrial apoptosis in LNCaP human prostate cancer cells. Exp Biol Med (Maywood) 2005;230:171-9.
248. Ivanov NI, Cowell SP, Brown P, Rennie PS, Guns ES, Cox ME. Lycopene differentially induces quiescence and apoptosis in androgen-responsive and -independent prostate cancer cell lines. Clin Nutr 2007;26:252-63.
249. Salman H, Bergman M, Djaldetti M, Bessler H. Lycopene affects proliferation and apoptosis of four malignant cell lines. Biomed Pharmacother 2007;61:366-9.
250. Cohn W, Thürmann P, Tenter U, Aebischer C, Schierle J, Schalch W. Comparative multiple dose plasma kinetics of lycopene administered in tomato juice, tomato soup or lycopene tablets. Eur J Nutr 2004;43:304-12.
251. Unlu NZ, Bohn T, Francis DM, Nagaraja HN, Clinton SK, Schwartz SJ. Lycopene from heat-induced cis-isomer-rich tomato sauce is more bioavailable than from all-trans-rich tomato sauce in human subjects. Br J Nutr 2007;98:140-6.
252. Boileau TW, Boileau AC, Erdman JW. Bioavailability of all-trans and cis-isomers of lycopene. Exp Biol Med (Maywood) 2002;227:914-9.
253. Hadley CW, Miller EC, Schwartz SJ, Clinton SK. Tomatoes, lycopene, and prostate cancer: progress and promise. Exp Biol Med (Maywood) 2002;227:869-80.
254. Teodoro AJ, Perrone D, Martucci RB, Borojevic R. Lycopene isomerisation and storage in an in vitro model of murine hepatic stellate cells. Eur J Nutr 2009;48:261-8.
255. Tang GW, Wang XD, Russell RM, Krinsky NI. Characterization of beta-apo-13-carotenone and beta-apo-14’-carotenal as enzymatic products of the excentric cleavage of beta-carotene. Biochemistry 1991;30:9829-34.
256. Wang XD, Tang GW, Fox JG, Krinsky NI, Russell RM. Enzymatic conversion of beta-carotene into beta-apo-carotenals and retinoids by human, monkey, ferret, and rat tissues. Arch Biochem Biophys 1991;285:8-16.
257. Kiefer C, Hessel S, Lampert JM, et al. Identification and characterization of a mammalian enzyme catalyzing the asymmetric oxidative cleavage of provitamin A. J Biol Chem 2001;276:14110-6.
258. Hu KQ, Liu C, Ernst H, et al. The biochemical characterization of ferret carotene-9’,10’-monooxygenase catalyzing cleavage of carotenoids in vitro and in vivo. J Biol Chem 2006;281:19327-38.
259. Amengual J, Lobo GP, Golczak M, et al. A mitochondrial enzyme degrades carotenoids and protects against oxidative stress. FASEB J 2011;25:948-59.
260. Kopec RE, Riedl KM, Harrison EH, et al. Identification and quantification of apo-lycopenals in fruits, vegetables, and human plasma. J Agric Food Chem 2010;58:3290-6.
261. Mein JR, Dolnikowski GG, Ernst H, Russell RM, Wang XD. Enzymatic formation of apo-carotenoids from the xanthophyll carotenoids lutein, zeaxanthin and β-cryptoxanthin by ferret carotene-9’,10’-monooxygenase. Arch Biochem Biophys 2011;506:109-21.
262. Kucuk O, Sarkar FH, Sakr W, et al. Phase II randomized clinical trial of lycopene supplementation before radical prostatectomy. Cancer Epidemiol Biomarkers Prev 2001;10:861-8.
263. Giovannucci E, Ascherio A, Rimm EB, Stampfer MJ, Colditz GA, Willett WC. Intake of carotenoids and retinol in relation to risk of prostate cancer. J Natl Cancer Inst 1995;87:1767-76.
264. Lamartiniere CA, Moore JB, Brown NM, Thompson R, Hardin MJ, Barnes S. Genistein suppresses mammary cancer in rats. Carcinogenesis 1995;16:2833-40.
265. Radhakrishnan EK, Bava SV, Narayanan SS, et al. [6]-Gingerol induces caspase-dependent apoptosis and prevents PMA-induced proliferation in colon cancer cells by inhibiting MAPK/AP-1 signaling. PLoS One 2014;9:e104401.
266. Shan Y, Wei Z, Tao L, et al. Prophylaxis of diallyl disulfide on skin carcinogenic model via p21-dependent Nrf2 stabilization. Sci Rep 2016;6:35676.
267. Liu Y, Bi T, Wang G, et al. Lupeol inhibits proliferation and induces apoptosis of human pancreatic cancer PCNA-1 cells through AKT/ERK pathways. Naunyn Schmiedebergs Arch Pharmacol 2015;388:295-304.
268. Chilampalli C, Zhang X, Kaushik RS, et al. Chemopreventive effects of combination of honokiol and magnolol with α-santalol on skin cancer developments. Drug Discov Ther 2013;7:109-15.
269. Aziz MH, Dreckschmidt NE, Verma AK. Plumbagin, a medicinal plant-derived naphthoquinone, is a novel inhibitor of the growth and invasion of hormone-refractory prostate cancer. Cancer Res 2008;68:9024-32.
270. Vadhanam MV, Ravoori S, Aqil F, Gupta RC. Chemoprevention of mammary carcinogenesis by sustained systemic delivery of ellagic acid. Eur J Cancer Prev 2011;20:484-91.
271. Gibellini L, Pinti M, Nasi M, et al. Quercetin and cancer chemoprevention. Evid Based Complement Alternat Med 2011;2011:591356.
273. Wu S, Zhu W, Thompson P, Hannun YA. Evaluating intrinsic and non-intrinsic cancer risk factors. Nat Commun 2018;9:3490.
274. Mendez D, Gaulton A, Bento AP, et al. ChEMBL: towards direct deposition of bioassay data. Nucleic Acids Res 2019;47:D930-40.
275. Jensen LJ, Kuhn M, Stark M, et al. STRING 8--a global view on proteins and their functional interactions in 630 organisms. Nucleic Acids Res 2009;37:D412-6.
276. Pezzuto JM. Grapes and Health. Springer; 2016.
277. Sergides C, Chirilă M, Silvestro L, Pitta D, Pittas A. Bioavailability and safety study of resveratrol 500 mg tablets in healthy male and female volunteers. Exp Ther Med 2016;11:164-70.
278. van Eck NJ, Waltman L. Software survey: VOSviewer, a computer program for bibliometric mapping. Scientometrics 2010;84:523-38.
279. Hoshino J, Park EJ, Kondratyuk TP, et al. Selective synthesis and biological evaluation of sulfate-conjugated resveratrol metabolites. J Med Chem 2010;53:5033-43.
280. Wang P, Li D, Ke W, Liang D, Hu X, Chen F. Resveratrol-induced gut microbiota reduces obesity in high-fat diet-fed mice. Int J Obes (Lond) 2020;44:213-25.
281. Long-Smith C, O’Riordan KJ, Clarke G, Stanton C, Dinan TG, Cryan JF. Microbiota-gut-brain axis: new therapeutic opportunities. Annu Rev Pharmacol Toxicol 2020;60:477-502.
282. Miller KD, Nogueira L, Mariotto AB, et al. Cancer treatment and survivorship statistics, 2019. CA Cancer J Clin 2019;69:363-85.
283. Yi LT, Dong SQ, Wang SS, et al. Curcumin attenuates cognitive impairment by enhancing autophagy in chemotherapy. Neurobiol Dis 2020;136:104715.
284. Tong Y, Wang K, Sheng S, Cui J. Polydatin ameliorates chemotherapy-induced cognitive impairment (chemobrain) by inhibiting oxidative stress, inflammatory response, and apoptosis in rats. Biosci Biotechnol Biochem 2020;84:1201-10.
285. Sporn M. Foreword. Natural Products for Cancer Chemoprevention. Springer; 2020. pp. vii-viii.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Dave A, Parande F, Park EJ, Pezzuto JM. Phytochemicals and cancer chemoprevention. J Cancer Metastasis Treat 2020;6:46. http://dx.doi.org/10.20517/2394-4722.2020.106
AMA Style
Dave A, Parande F, Park EJ, Pezzuto JM. Phytochemicals and cancer chemoprevention. Journal of Cancer Metastasis and Treatment. 2020; 6: 46. http://dx.doi.org/10.20517/2394-4722.2020.106
Chicago/Turabian Style
Dave, Asim, Falguni Parande, Eun-Jung Park, John M. Pezzuto. 2020. "Phytochemicals and cancer chemoprevention" Journal of Cancer Metastasis and Treatment. 6: 46. http://dx.doi.org/10.20517/2394-4722.2020.106
ACS Style
Dave, A.; Parande F.; Park E.J.; Pezzuto JM. Phytochemicals and cancer chemoprevention. J. Cancer. Metastasis. Treat. 2020, 6, 46. http://dx.doi.org/10.20517/2394-4722.2020.106
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 57 clicks
Cite This Article 57 clicks

 Like This Article 4
likes
Like This Article 4
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.